

Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart
- PMID: 23204520
- PMCID: PMC3542989
- DOI: 10.1074/jbc.M112.426957
Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart
Abstract
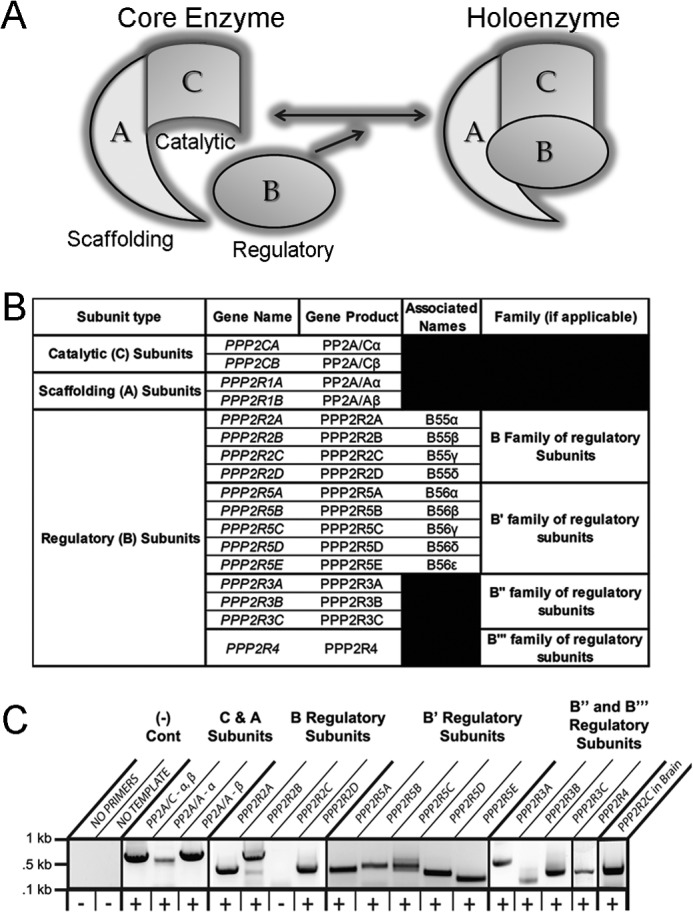
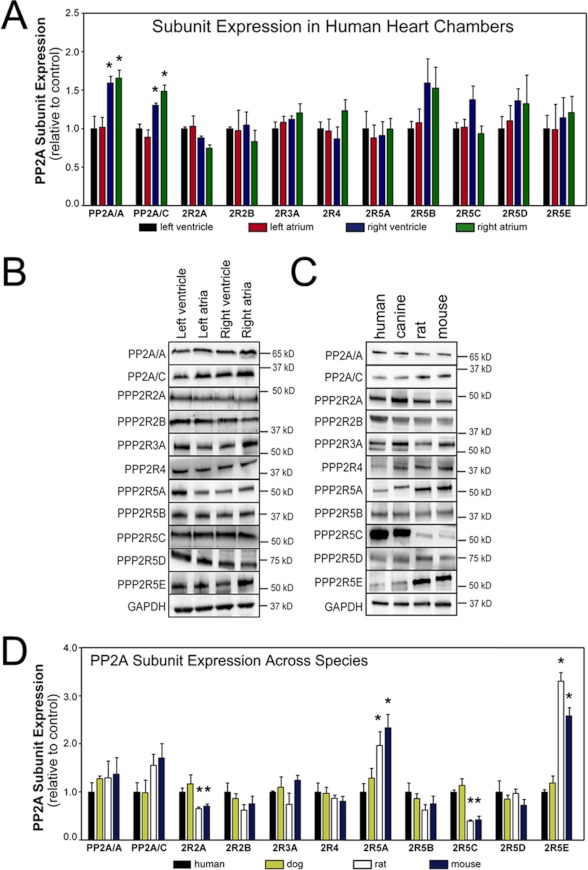
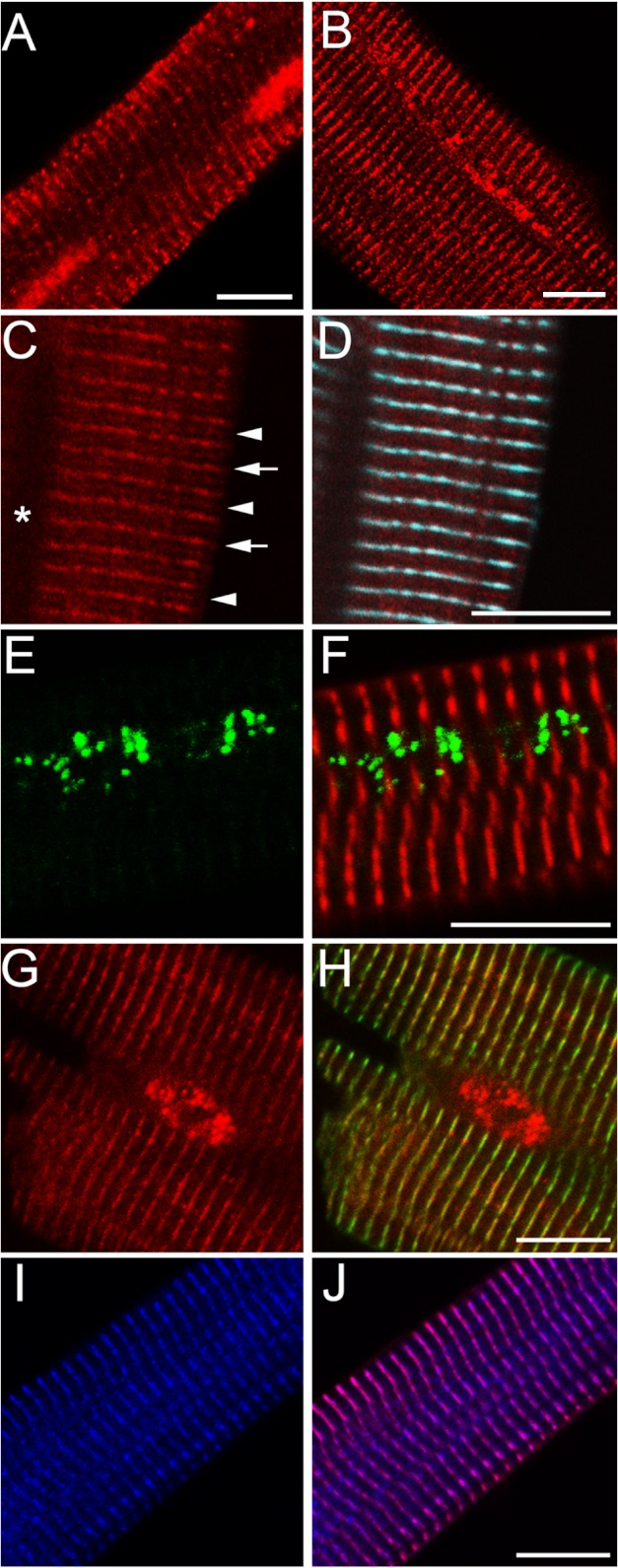
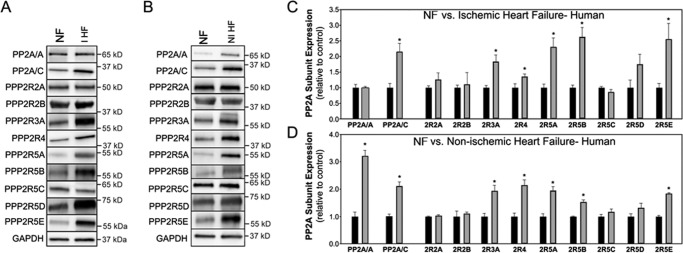
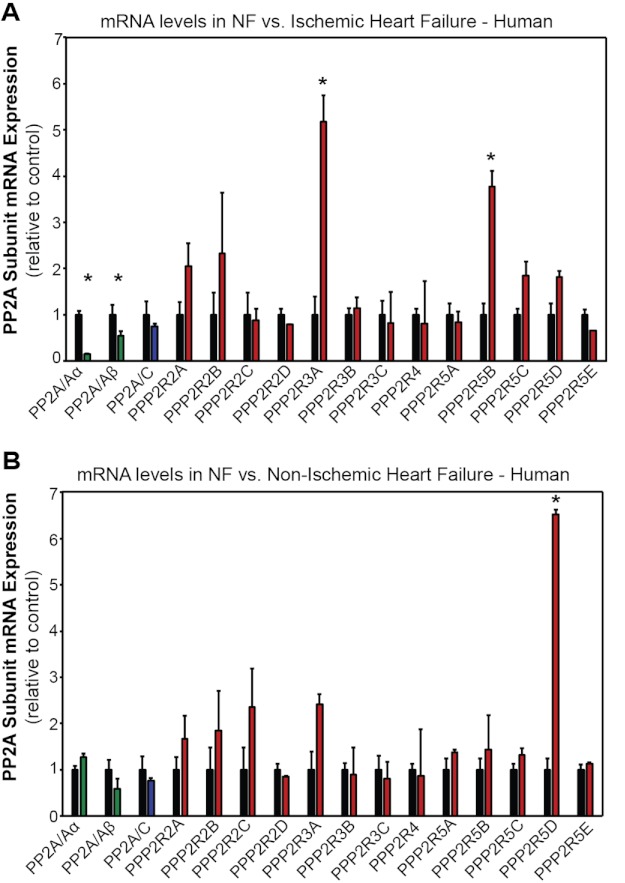
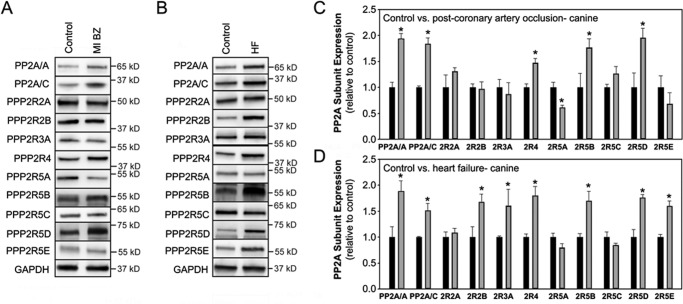
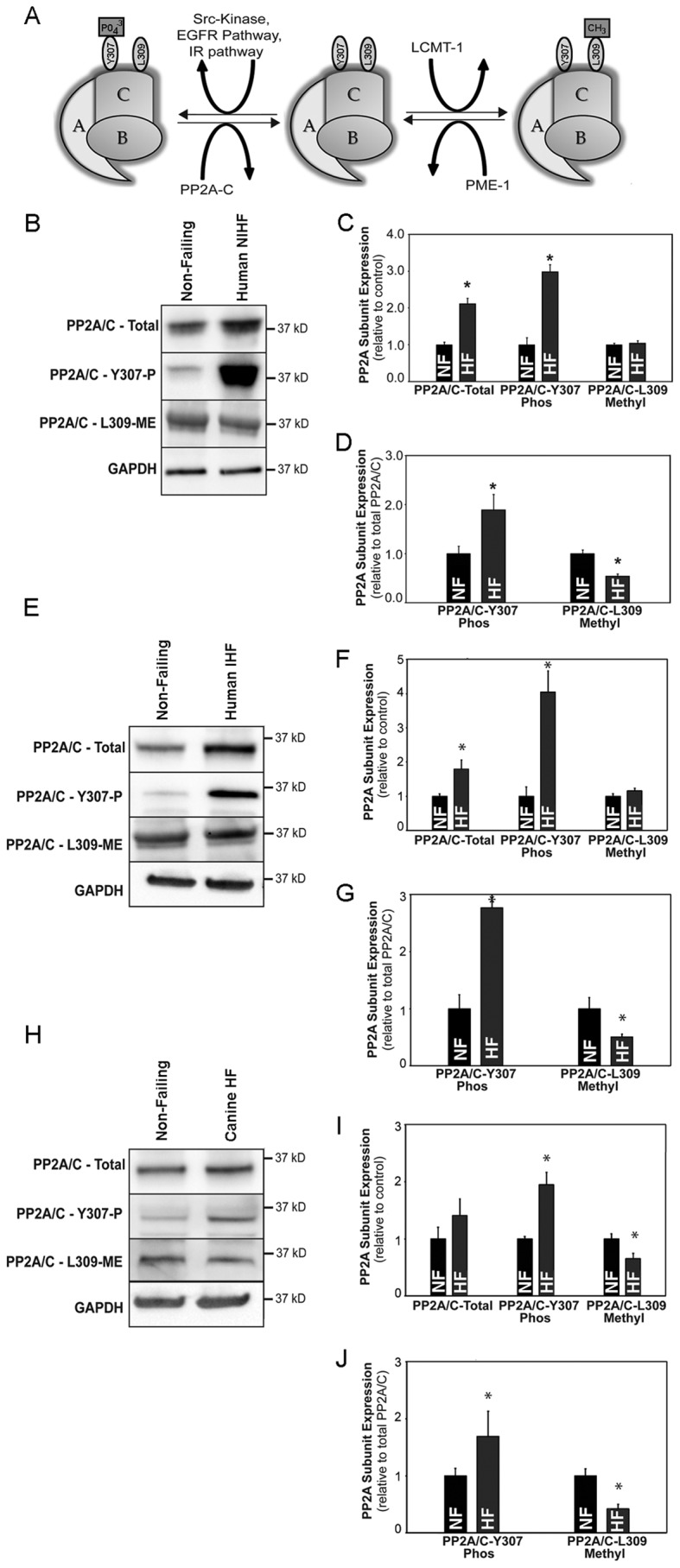
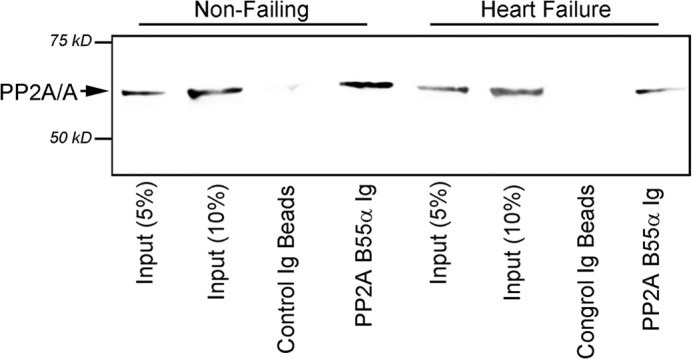
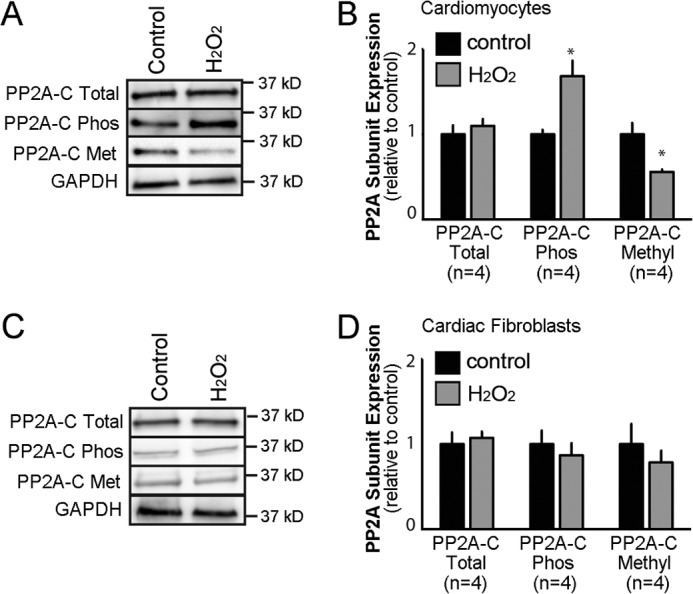
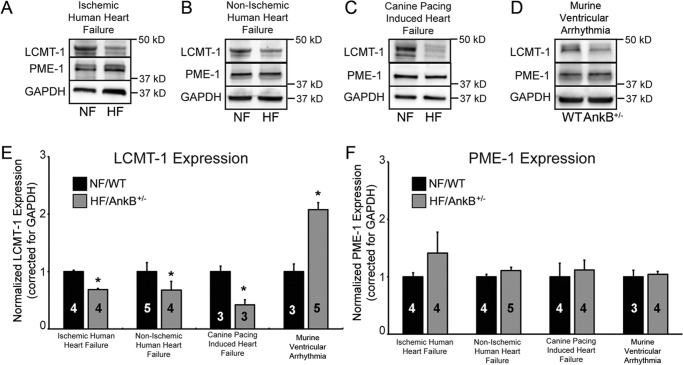
Kinase/phosphatase balance governs cardiac excitability in health and disease. Although detailed mechanisms for cardiac kinase regulation are established, far less is known regarding cardiac protein phosphatase 2A (PP2A) regulation. This is largely due to the complexity of the PP2A holoenzyme structure (combinatorial assembly of three subunit enzyme from >17 subunit genes) and the inability to segregate "global" PP2A function from the activities of multiple "local" holoenzyme populations. Here we report that PP2A catalytic, regulatory, and scaffolding subunits are tightly regulated at transcriptional, translational, and post-translational levels to tune myocyte function at base line and in disease. We show that past global read-outs of cellular PP2A activity more appropriately represent the collective activity of numerous individual PP2A holoenzymes, each displaying a specific subcellular localization (dictated by select PP2A regulatory subunits) as well as local specific post-translational catalytic subunit methylation and phosphorylation events that regulate local and rapid holoenzyme assembly/disassembly (via leucine carboxymethyltransferase 1/phosphatase methylesterase 1 (LCMT-1/PME-1). We report that PP2A subunits are selectively regulated between human and animal models, across cardiac chambers, and even within specific cardiac cell types. Moreover, this regulation can be rapidly tuned in response to cellular activation. Finally, we report that global PP2A is altered in human and experimental models of heart disease, yet each pathology displays its own distinct molecular signature though specific PP2A subunit modulatory events. These new data provide an initial view into the signaling pathways that govern PP2A function in heart but also establish the first step in defining specific PP2A regulatory targets in health and disease.
Figures
References
-
- He B. J., Joiner M. L., Singh M. V., Luczak E. D., Swaminathan P. D., Koval O. M., Kutschke W., Allamargot C., Yang J., Guan X., Zimmerman K., Grumbach I. M., Weiss R. M., Spitz D. R., Sigmund C. D., Blankesteijn W. M., Heymans S., Mohler P. J., Anderson M. E. (2011) Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 17, 1610–1618 - PMC - PubMed
-
- Chelu M. G., Sarma S., Sood S., Wang S., van Oort R. J., Skapura D. G., Li N., Santonastasi M., Müller F. U., Schmitz W., Schotten U., Anderson M. E., Valderrábano M., Dobrev D., Wehrens X. H. (2009) Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951 - PMC - PubMed
-
- Swaminathan P. D., Purohit A., Soni S., Voigt N., Singh M. V., Glukhov A. V., Gao Z., He B. J., Luczak E. D., Joiner M. L., Kutschke W., Yang J., Donahue J. K., Weiss R. M., Grumbach I. M., Ogawa M., Chen P. S., Efimov I., Dobrev D., Mohler P. J., Hund T. J., Anderson M. E. (2011) Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Invest. 121, 3277–3288 - PMC - PubMed
-
- Reiken S., Gaburjakova M., Gaburjakova J., He Kl K. L., Prieto A., Becker E., Yi Gh G. H., Wang J., Burkhoff D., Marks A. R. (2001) β-Adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation 104, 2843–2848 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL084583/HL/NHLBI NIH HHS/United States
- R01 HL079031/HL/NHLBI NIH HHS/United States
- HL70250/HL/NHLBI NIH HHS/United States
- R01 HL096652/HL/NHLBI NIH HHS/United States
- R01 HL084583/HL/NHLBI NIH HHS/United States
- R01 HL113001/HL/NHLBI NIH HHS/United States
- R01 HL070250/HL/NHLBI NIH HHS/United States
- R00 HL096805/HL/NHLBI NIH HHS/United States
- HL114893/HL/NHLBI NIH HHS/United States
- HL62494/HL/NHLBI NIH HHS/United States
- R01 HL062494/HL/NHLBI NIH HHS/United States
- K99 HL096805/HL/NHLBI NIH HHS/United States
- HL089836/HL/NHLBI NIH HHS/United States
- R01 HL083422/HL/NHLBI NIH HHS/United States
- HL083422/HL/NHLBI NIH HHS/United States
- HL079031/HL/NHLBI NIH HHS/United States
- HL066140/HL/NHLBI NIH HHS/United States
- HL096805/HL/NHLBI NIH HHS/United States
- R01 HL089836/HL/NHLBI NIH HHS/United States
- T32 GM007337/GM/NIGMS NIH HHS/United States
- R01 HL066140/HL/NHLBI NIH HHS/United States
- R01 HL114893/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
