

Pathophysiology and Clinical Manifestations of the β-Thalassemias
- PMID: 23209183
- PMCID: PMC3543079
- DOI: 10.1101/cshperspect.a011726
Pathophysiology and Clinical Manifestations of the β-Thalassemias
Abstract
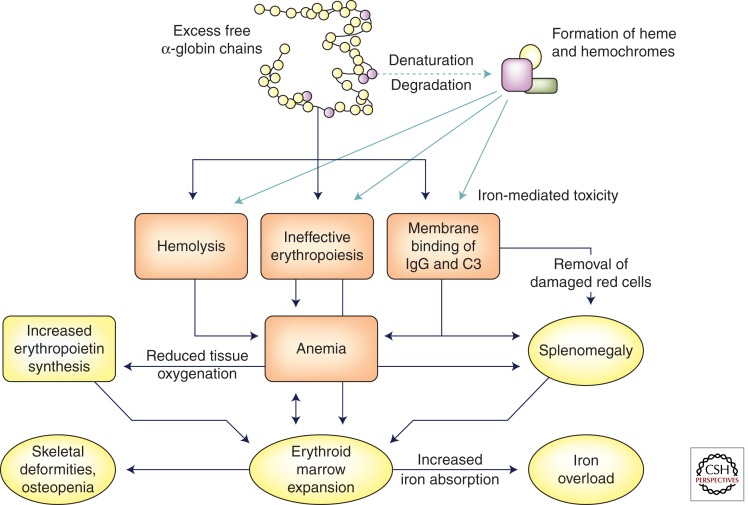
The β-thalassemia syndromes reflect deficient or absent β-globin synthesis usually owing to a mutation in the β-globin locus. The relative excess of α-globin results in the formation of insoluble aggregates leading to ineffective erythropoiesis and shortened red cell survival. A relatively high capacity for fetal hemoglobin synthesis is a major genetic modifier of disease severity, with polymorphisms in other genes also having a significant role. Iron overload secondary to enhanced absorption and red cell transfusions causes an increase in liver iron and in various other tissues, leading to endocrine and cardiac dysfunction. Modern chelation regimens are effective in removing iron and preserving or restoring organ function.
Figures
References
-
- Anderson LJ, Westwood MA, Holden S, Davis B, Prescott E, Wonke B, Porter JB, Walker JM, Pennell DJ 2004. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: A prospective study using T2* cardiovascular magnetic resonance. Br J Haematol 127: 348–355 - PubMed
-
- Bank A, Marks PA 1966. Excess α chain synthesis relative to β chain synthesis in thalassaemia major and minor. Nature 212: 1198–1200 - PubMed
-
- Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, Romeo MA, Forni GL, Gamberini MR, Ghilardi R, et al. 2004. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 89: 1187–1193 - PubMed
-
- Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Fomi GL, Gamberini MR, Ghilard R, Piga A, Romeo MA, Zhao H, et al. 2006. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood 107: 3733–3737 - PubMed
-
- Borgna-Pignatti C, Marsella M, Zanforrin N 2010. The natural history of thalassemia intermedia. Ann NY Acad Sci 1202: 214–220 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources