

Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons
- PMID: 23212910
- PMCID: PMC3554895
- DOI: 10.1074/jbc.M112.391680
Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons
Abstract
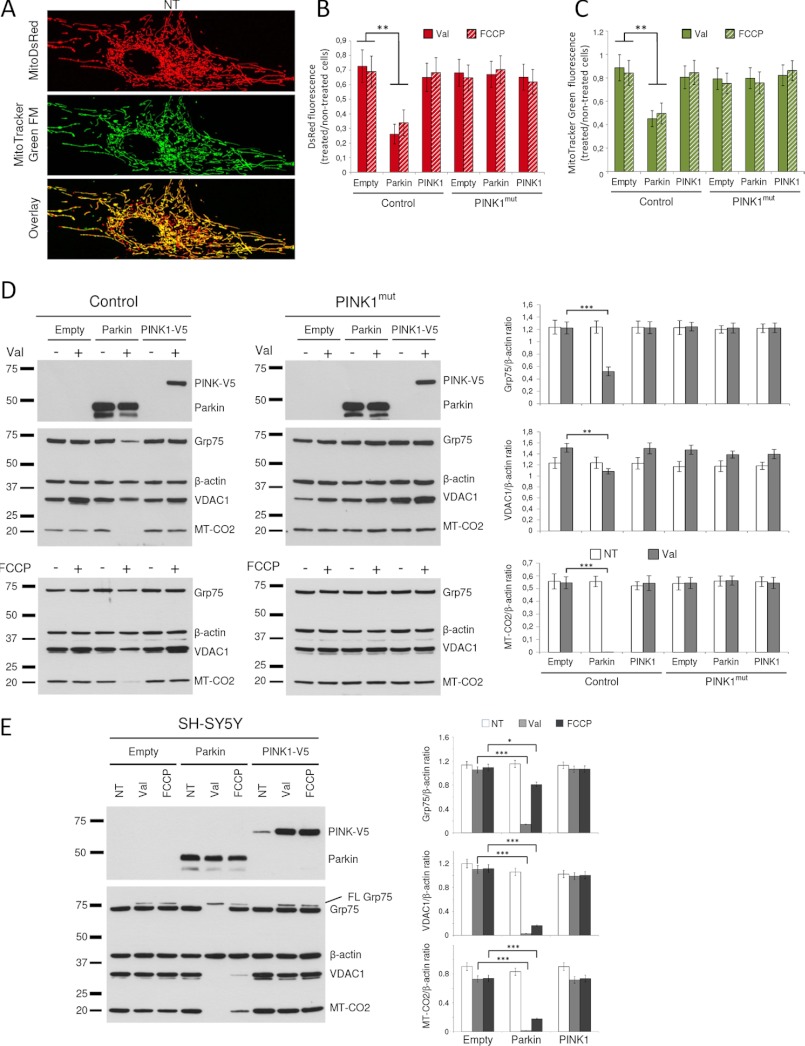
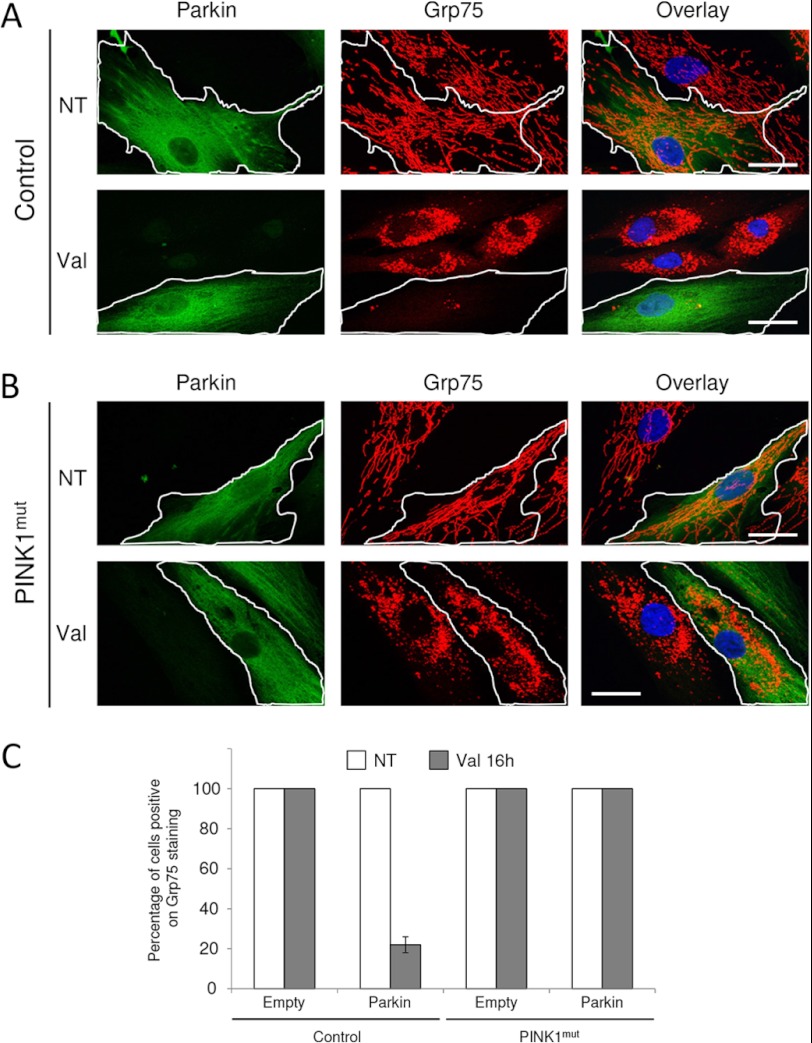
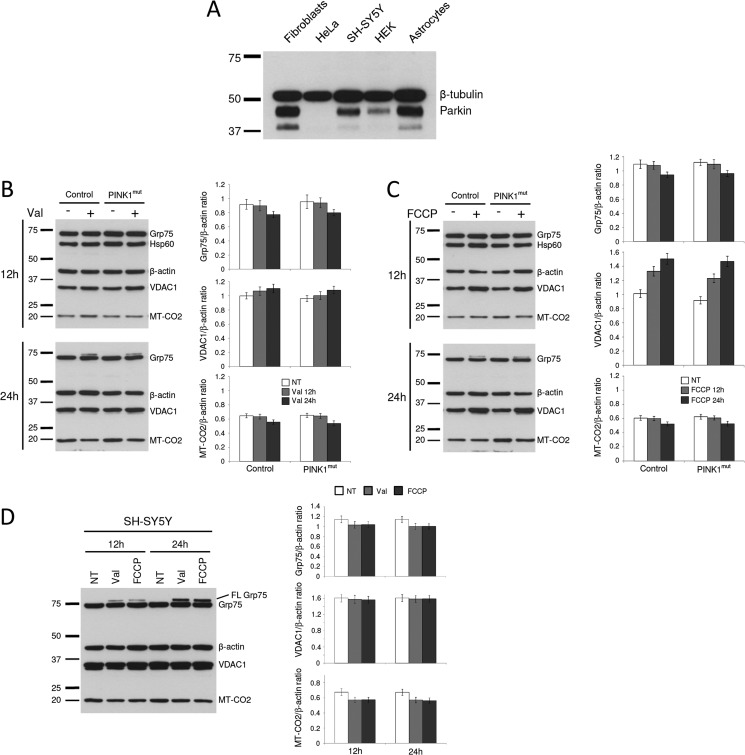
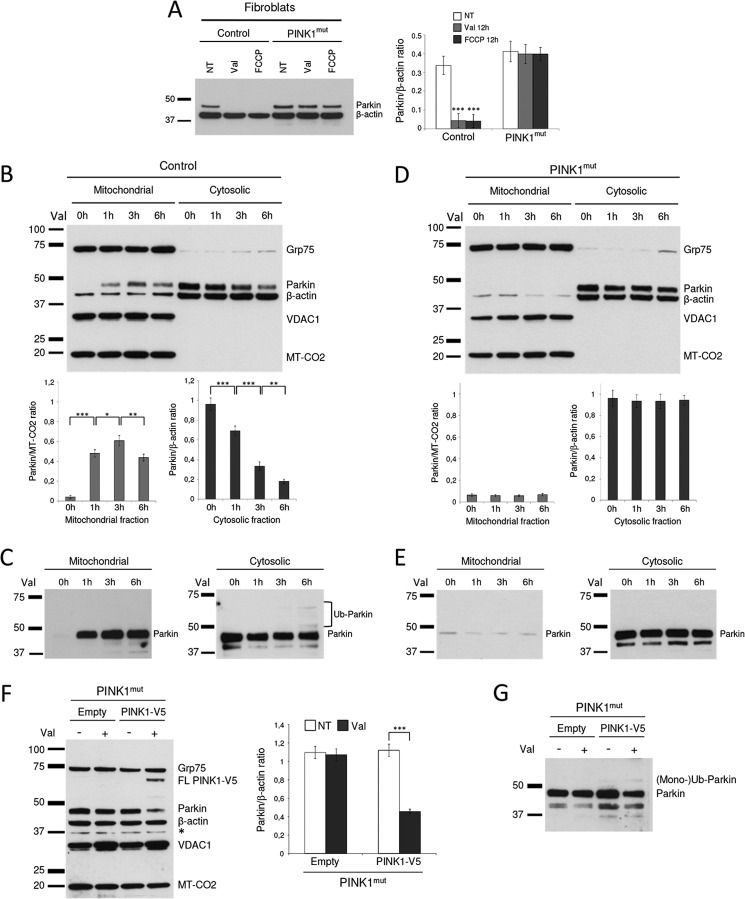
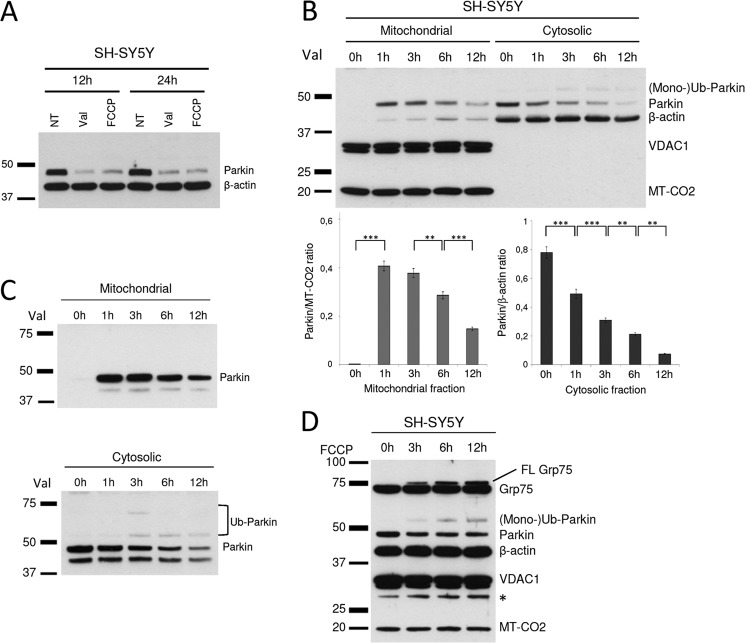
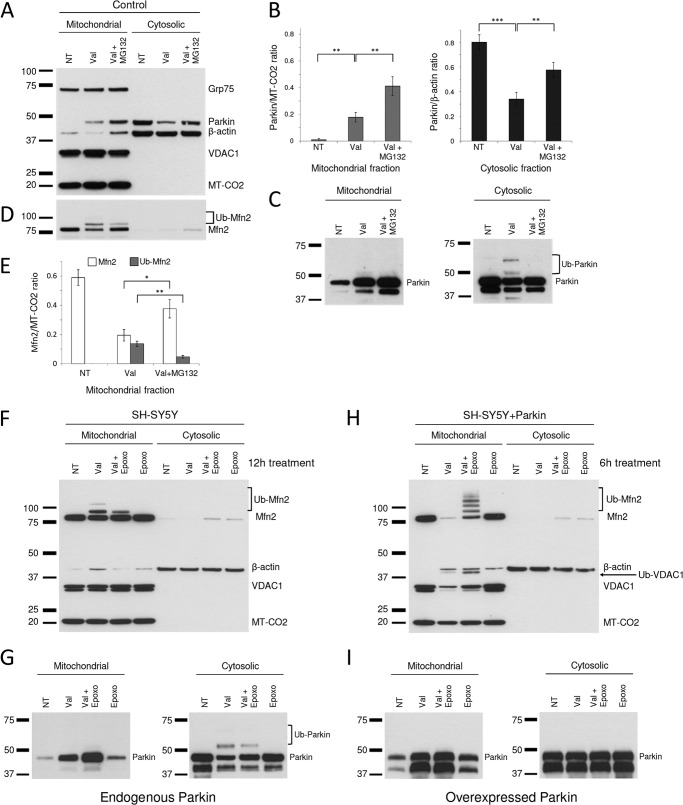
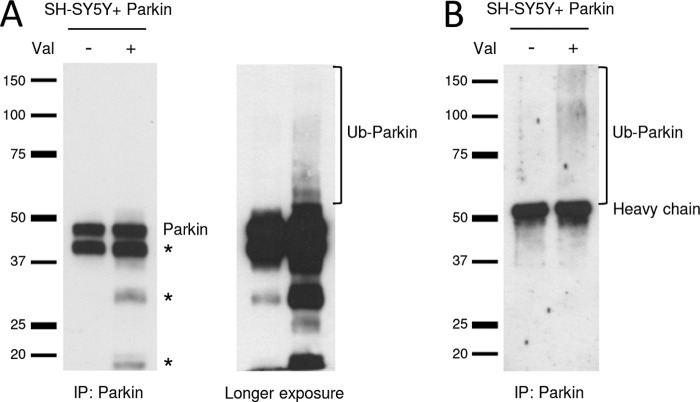
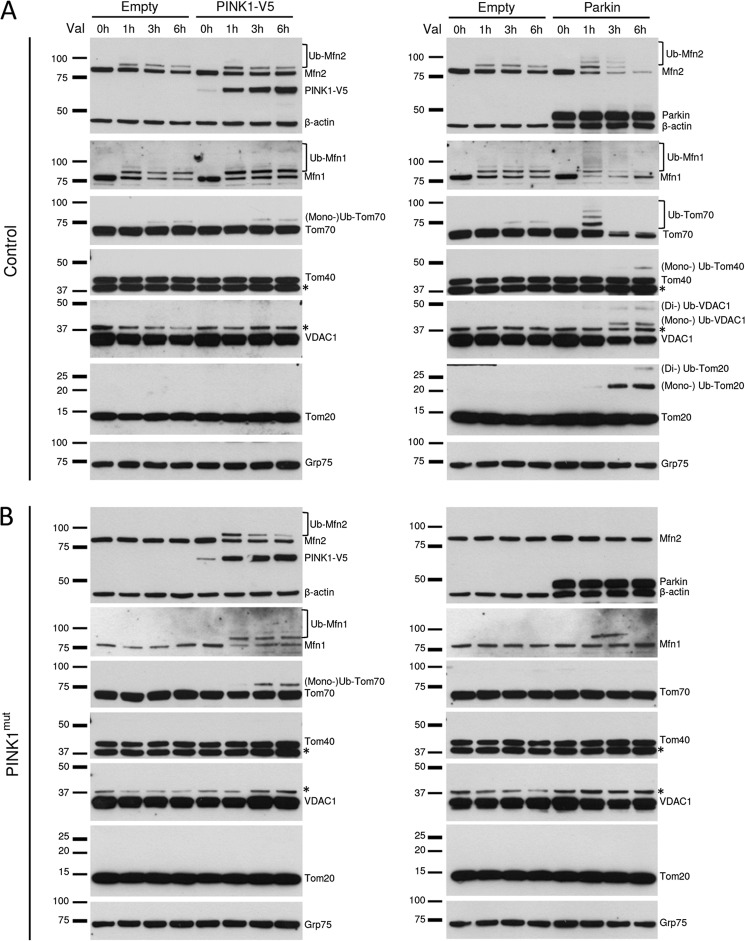
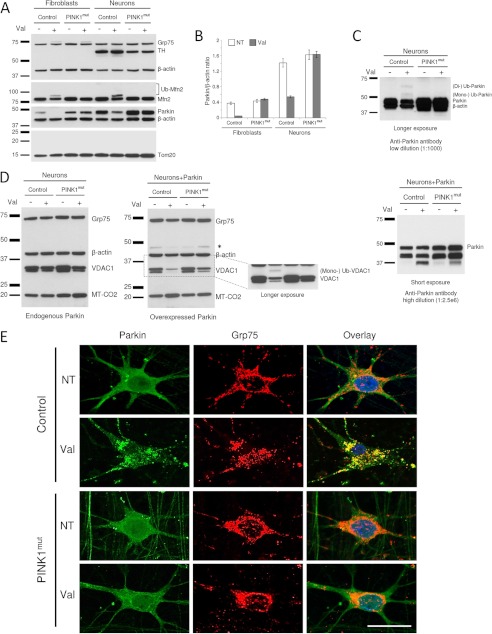
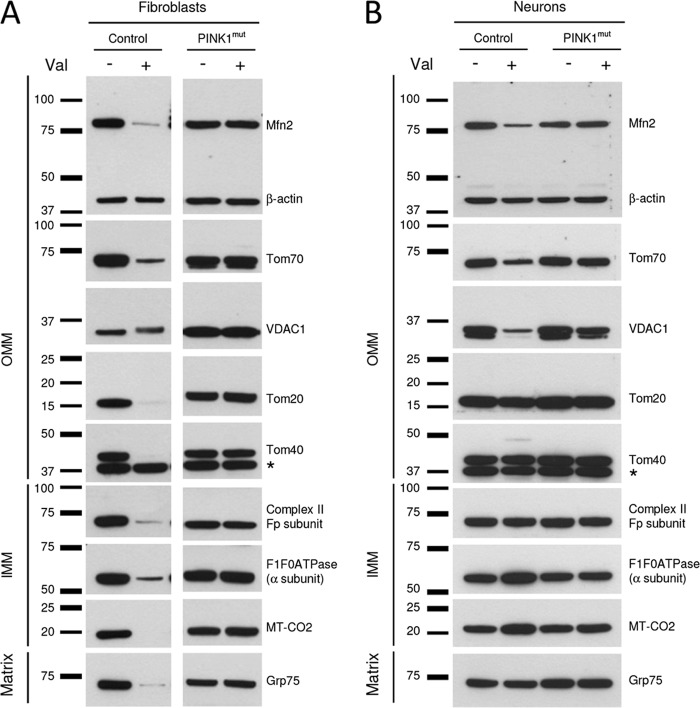
Mutations in the E3 ubiquitin ligase Parkin and the mitochondrial PTEN-induced putative kinase 1 (PINK1) have been identified to cause autosomal recessive forms of familial Parkinson disease, with PINK1 functioning upstream of Parkin in a pathway important for the maintenance of mitochondrial function and morphology. Upon the loss of the mitochondrial membrane potential, Parkin translocates to mitochondria in a PINK1-dependent manner to ubiquitinate mitochondrial proteins. Parkin-mediated polyubiquitination of outer mitochondrial membrane (OMM) proteins recruits the ubiquitin- and LC3-binding adaptor protein p62 to mitochondria and induces mitophagy. Although previous studies examined mitophagy in established cell lines through overexpression approaches, there is an imperative to study the role of endogenous Parkin and PINK1 in human-derived and biologically relevant cell models. Here, we demonstrate in human primary fibroblasts and induced pluripotent stem-derived neurons from controls and PINK1 mutation carriers that endogenous levels of Parkin are not sufficient to initiate mitophagy upon loss of the mitochondrial membrane potential, caused by its (self-)ubiquitination, followed by degradation via the ubiquitin proteasome system. Next, we showed differential PINK1-dependent, Parkin-mediated ubiquitination of OMM proteins, which is Parkin dose-dependent and affects primarily OMM proteins of higher molecular mass. In contrast to the situation fibroblasts, we did not detect mitophagy in induced pluripotent stem-derived neurons even upon overexpression of Parkin. Taken together, our data demonstrate that mitophagy differs between human non-neuronal and neuronal cells and between "endogenous" and "Parkin-overexpressing" cellular models.
Figures
References
-
- Langston J. W., Ballard P., Tetrud J. W., Irwin I. (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 - PubMed
-
- Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., Greenamyre J. T. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3, 1301–1306 - PubMed
-
- Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 - PubMed
-
- Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 - PubMed
-
- Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. (2006) Drosophila PINK1 is required for mitochondrial function and interacts genetically with Parkin. Nature 441, 1162–1166 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
