

ISG15 deregulates autophagy in genotoxin-treated ataxia telangiectasia cells
- PMID: 23212917
- PMCID: PMC3554909
- DOI: 10.1074/jbc.M112.403832
ISG15 deregulates autophagy in genotoxin-treated ataxia telangiectasia cells
Abstract
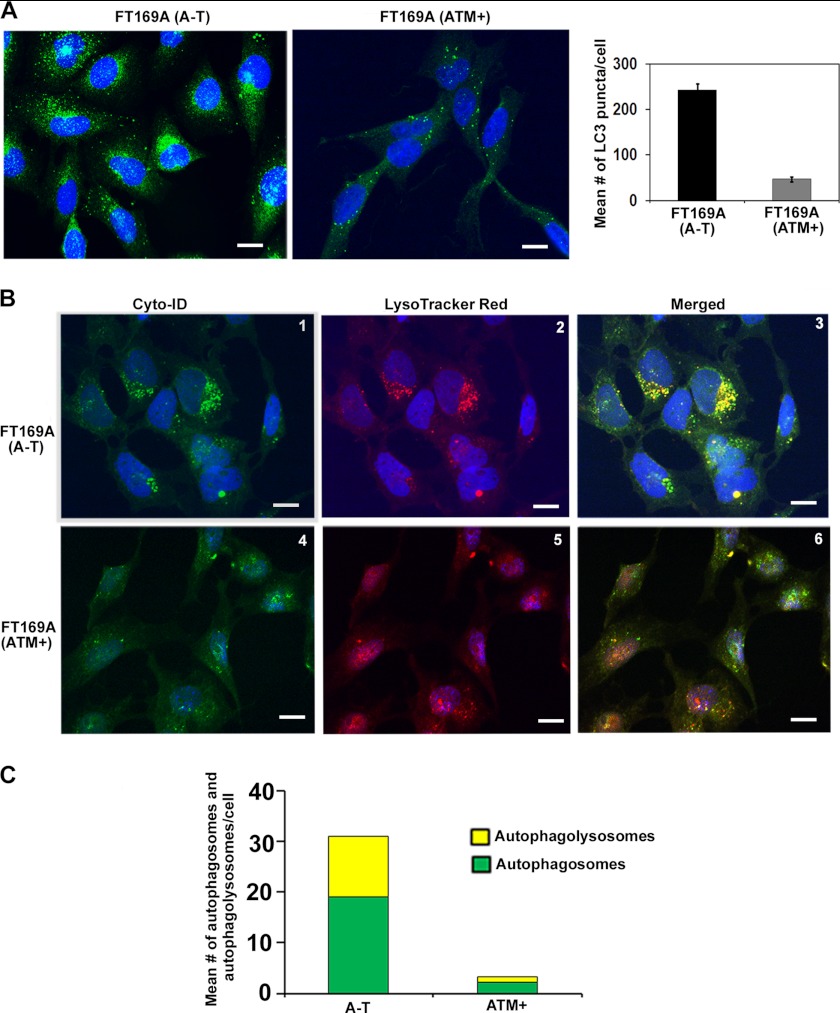
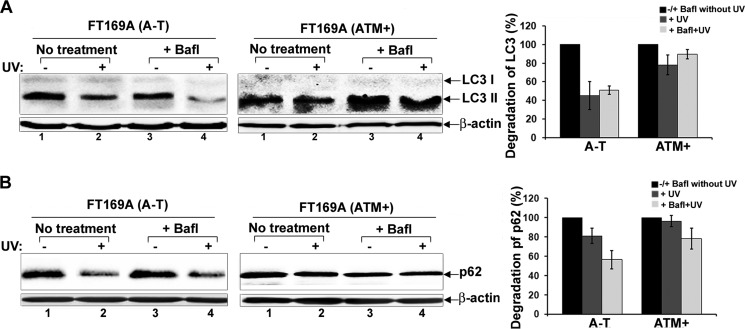
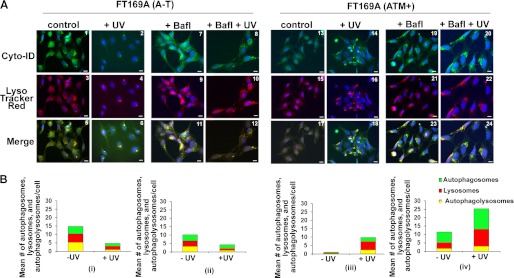
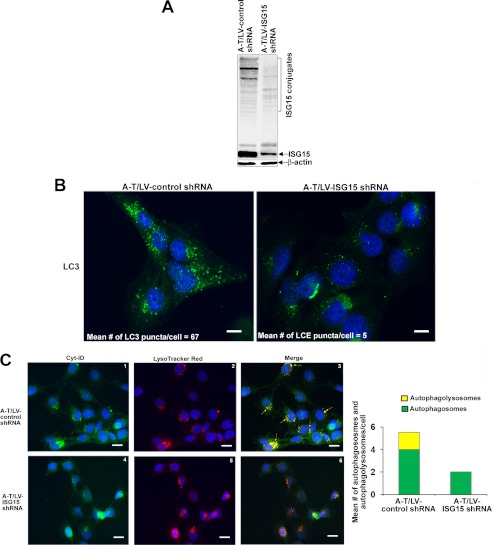
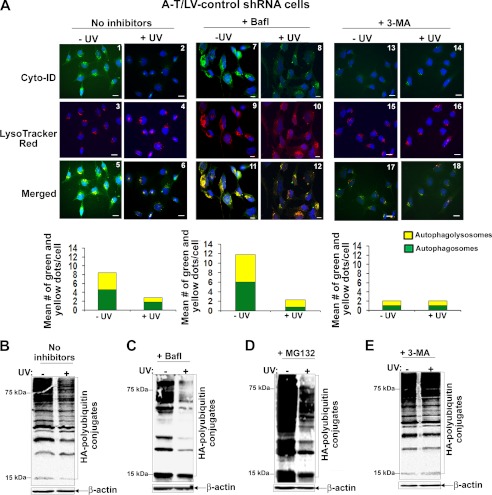
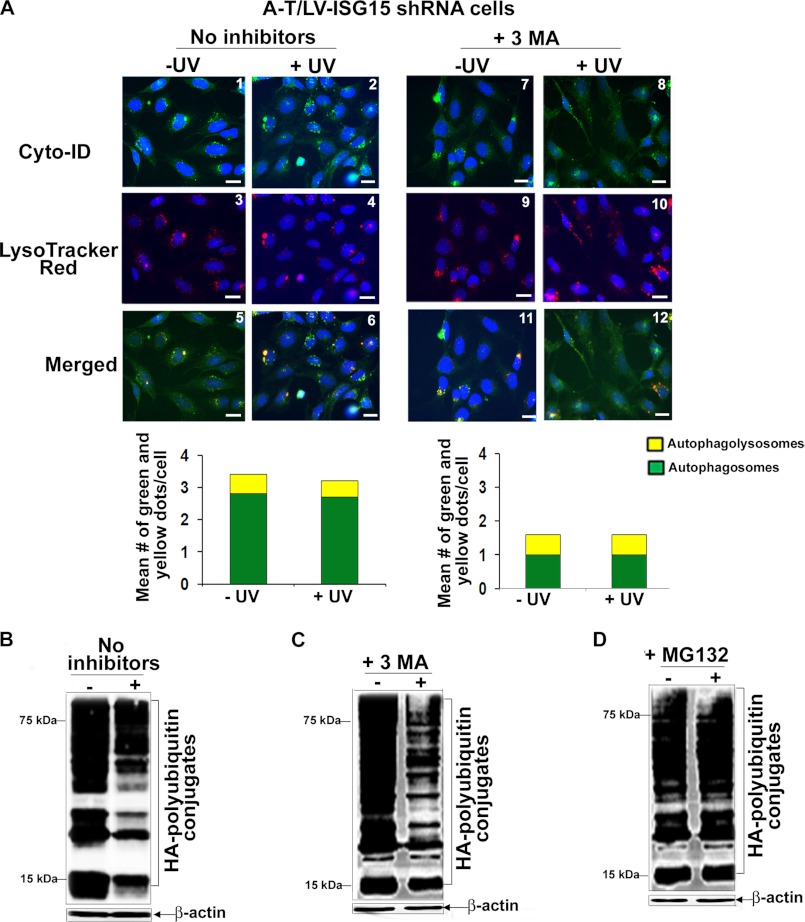
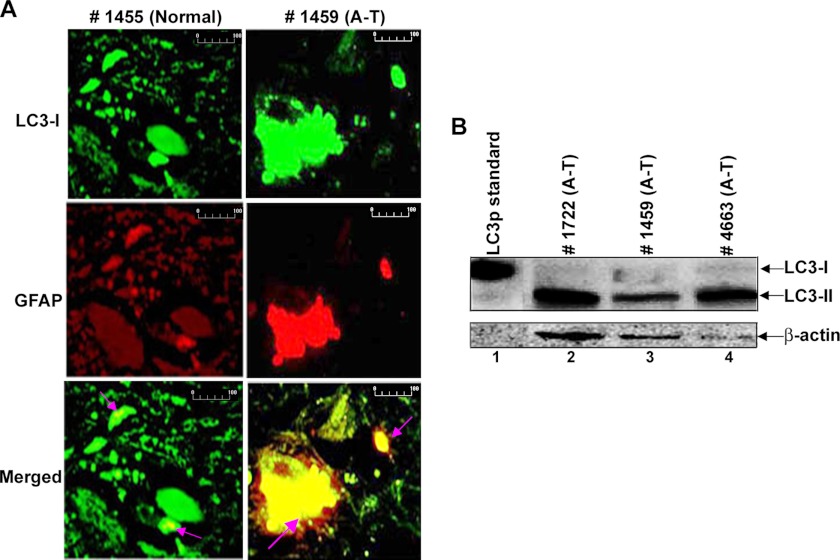
Ataxia-telangiectasia (A-T) is a cerebellar neurodegenerative disorder; however, the basis for the neurodegeneration in A-T is not well established. Lesions in the ubiquitin and autophagy pathways are speculated to contribute to the neurodegeneration in other neurological diseases and may have a role in A-T neurodegeneration. Our recent studies revealed that the constitutively elevated ISG15 pathway impairs targeted proteasome-mediated protein degradation in A-T cells. Here, we demonstrate that the basal autophagy pathway is activated in the ubiquitin pathway-compromised A-T cells. We also show that genotoxic stress triggers aberrant degradation of the proteasome and autophagy substrates (autophagic flux) in A-T cells. Inhibition of autophagy at an early stage using 3-methyladenine blocked UV-induced autophagic flux in A-T cells. On the other hand, bafilomycin A1, which inhibits autophagy at a late stage, failed to block UV-induced autophagic flux, suggesting that overinduction of autophagy may underlie aberrant autophagic flux in A-T cells. The ISG15-specific shRNA that restored proteasome function restores autophagic function in A-T cells. These findings suggest that autophagy compensates for the ISG15-dependent ablation of proteasome-mediated protein degradation in A-T cells. Genotoxic stress overactivates this compensatory mechanism, triggering aberrant autophagic flux in A-T cells. Supporting the model, we show that autophagy is activated in the brain tissues of human A-T patients. This highlights a plausible causal contribution of a novel "ISG15 proteinopathy" in A-T neuronal cell death.
Figures


References
-
- Frappart P. O., McKinnon P. J. (2006) Ataxia-telangiectasia and related diseases. Neuromolecular Med. 8, 495–511 - PubMed
-
- Lavin M. F., Khanna K. K. (1999) ATM. The protein encoded by the gene mutated in the radiosensitive syndrome ataxia-telangiectasia. Int. J. Radiat. Biol. 75, 1201–1214 - PubMed
-
- Boder E. (1985) Ataxia-telangiectasia. An overview. Kroc Found. Ser. 19, 1–63 - PubMed
-
- Chun H. H., Gatti R. A. (2004) Ataxia-telangiectasia, an evolving phenotype. DNA Repair 3, 1187–1196 - PubMed
-
- Easton D. F. (1994) Cancer risks in A-T heterozygotes. Int. J. Radiat. Biol. 66, S177–S182 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
