

Transition paths, diffusive processes, and preequilibria of protein folding
- PMID: 23213246
- PMCID: PMC3529084
- DOI: 10.1073/pnas.1209891109
Transition paths, diffusive processes, and preequilibria of protein folding
Abstract
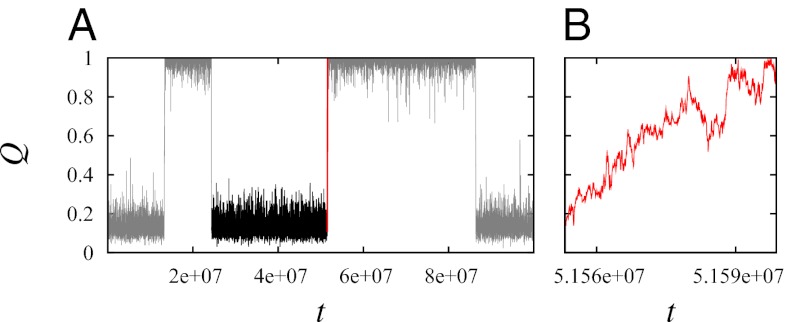
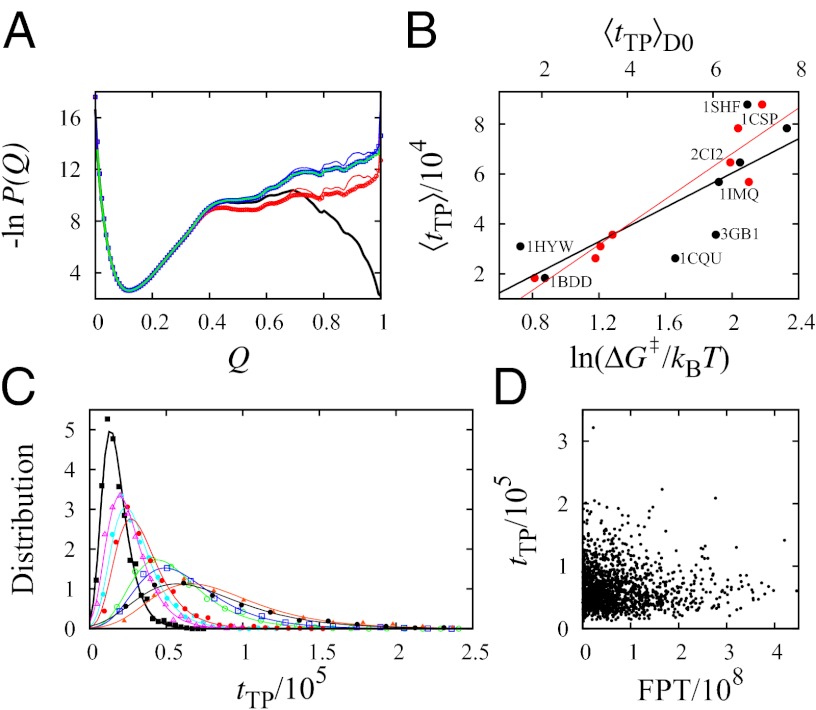
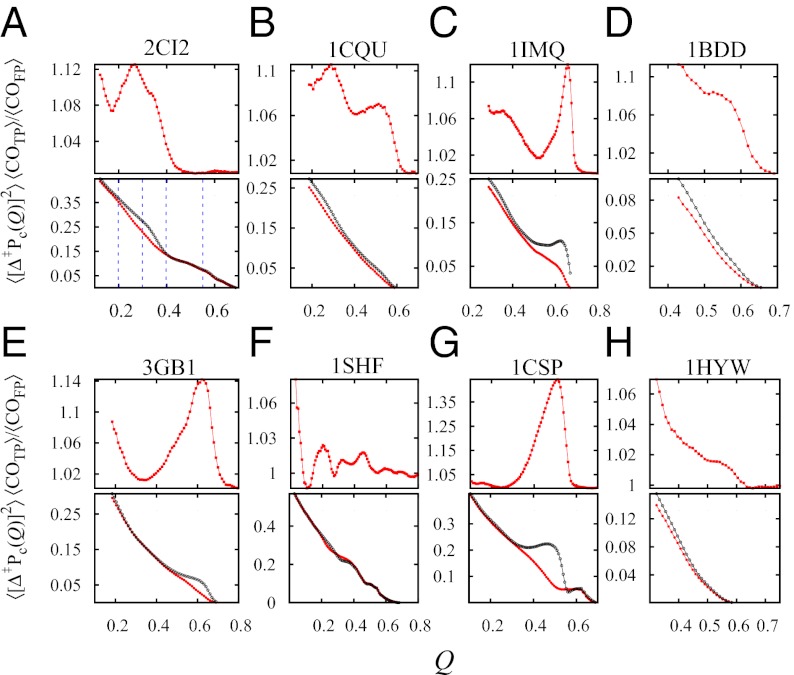
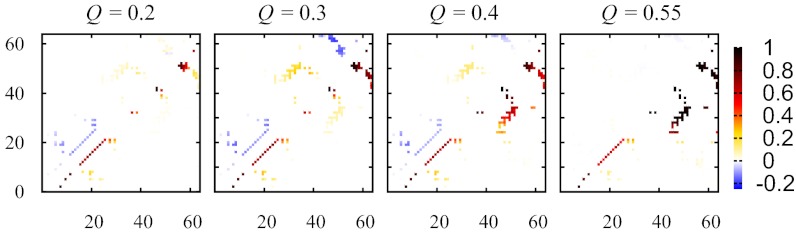
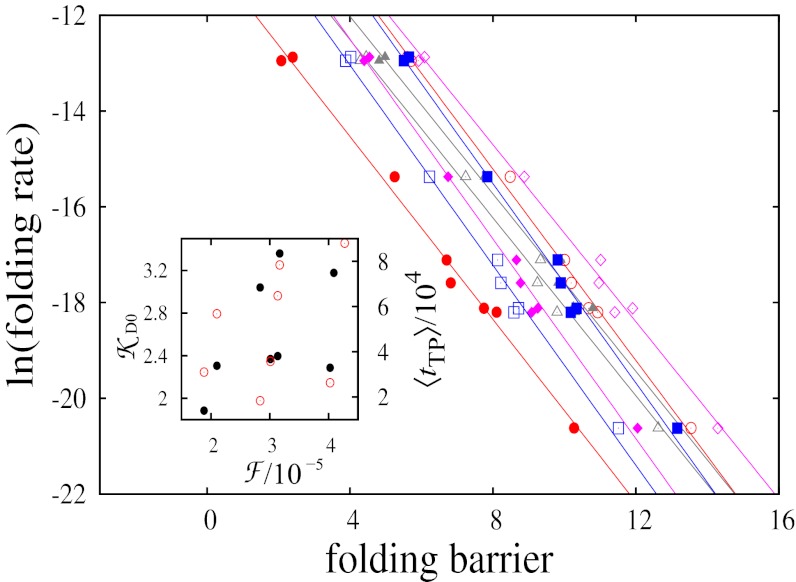
Fundamental relationships between the thermodynamics and kinetics of protein folding were investigated using chain models of natural proteins with diverse folding rates by extensive comparisons between the distribution of conformations in thermodynamic equilibrium and the distribution of conformations sampled along folding trajectories. Consistent with theory and single-molecule experiment, duration of the folding transition paths exhibits only a weak correlation with overall folding time. Conformational distributions of folding trajectories near the overall thermodynamic folding/unfolding barrier show significant deviations from preequilibrium. These deviations, the distribution of transition path times, and the variation of mean transition path time for different proteins can all be rationalized by a diffusive process that we modeled using simple Monte Carlo algorithms with an effective coordinate-independent diffusion coefficient. Conformations in the initial stages of transition paths tend to form more nonlocal contacts than typical conformations with the same number of native contacts. This statistical bias, which is indicative of preferred folding pathways, should be amenable to future single-molecule measurements. We found that the preexponential factor defined in the transition state theory of folding varies from protein to protein and that this variation can be rationalized by our Monte Carlo diffusion model. Thus, protein folding physics is different in certain fundamental respects from the physics envisioned by a simple transition-state picture. Nonetheless, transition state theory can be a useful approximate predictor of cooperative folding speed, because the height of the overall folding barrier is apparently a proxy for related rate-determining physical properties.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ikai A, Tanford C. Kinetic evidence for incorrectly folded intermediate states in the refolding of denatured proteins. Nature. 1971;230(5289):100–102. - PubMed
-
- Eyring H. The activated complex in chemical reactions. J Chem Phys. 1935;3:107–115.
-
- Matthews CR. Effect of point mutations on the folding of globular proteins. Methods Enzymol. 1987;154:498–511. - PubMed
-
- Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224(3):771–782. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
