

Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing
- PMID: 23217326
- PMCID: PMC3516590
- DOI: 10.1016/j.ajhg.2012.10.015
Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing
Abstract
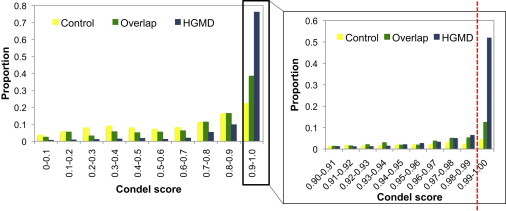
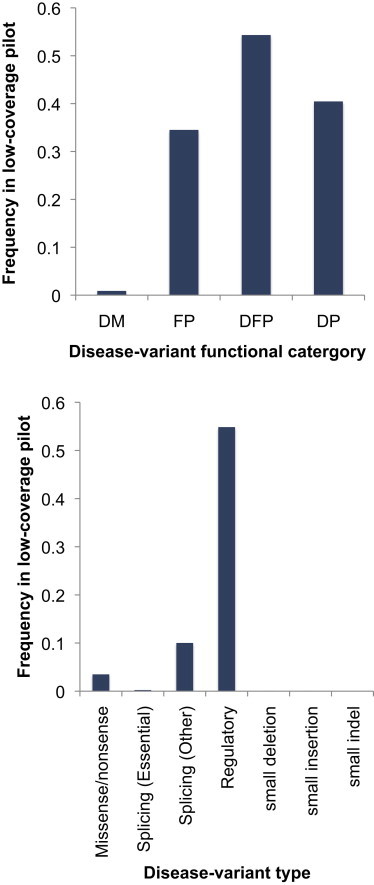
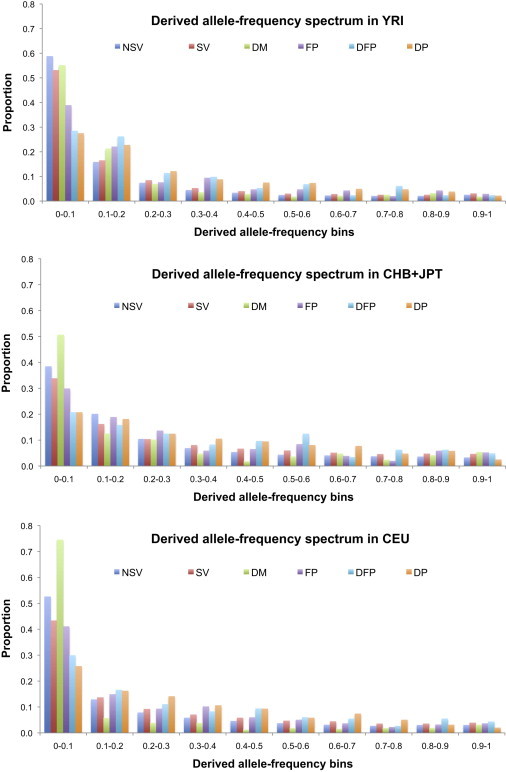
We have assessed the numbers of potentially deleterious variants in the genomes of apparently healthy humans by using (1) low-coverage whole-genome sequence data from 179 individuals in the 1000 Genomes Pilot Project and (2) current predictions and databases of deleterious variants. Each individual carried 281-515 missense substitutions, 40-85 of which were homozygous, predicted to be highly damaging. They also carried 40-110 variants classified by the Human Gene Mutation Database (HGMD) as disease-causing mutations (DMs), 3-24 variants in the homozygous state, and many polymorphisms putatively associated with disease. Whereas many of these DMs are likely to represent disease-allele-annotation errors, between 0 and 8 DMs (0-1 homozygous) per individual are predicted to be highly damaging, and some of them provide information of medical relevance. These analyses emphasize the need for improved annotation of disease alleles both in mutation databases and in the primary literature; some HGMD mutation data have been recategorized on the basis of the present findings, an iterative process that is both necessary and ongoing. Our estimates of deleterious-allele numbers are likely to be subject to both overcounting and undercounting. However, our current best mean estimates of ~400 damaging variants and ~2 bona fide disease mutations per individual are likely to increase rather than decrease as sequencing studies ascertain rare variants more effectively and as additional disease alleles are discovered.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
