

Cowchock syndrome is associated with a mutation in apoptosis-inducing factor
- PMID: 23217327
- PMCID: PMC3516602
- DOI: 10.1016/j.ajhg.2012.10.008
Cowchock syndrome is associated with a mutation in apoptosis-inducing factor
Abstract
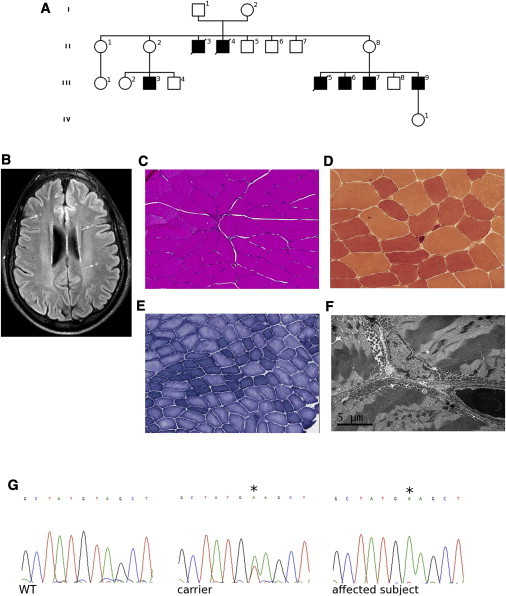
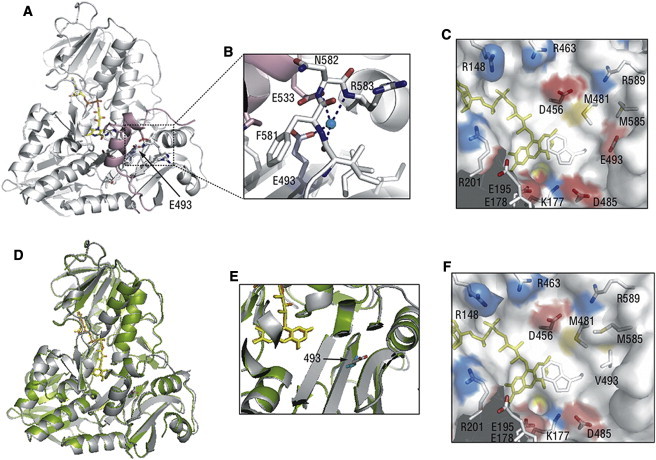
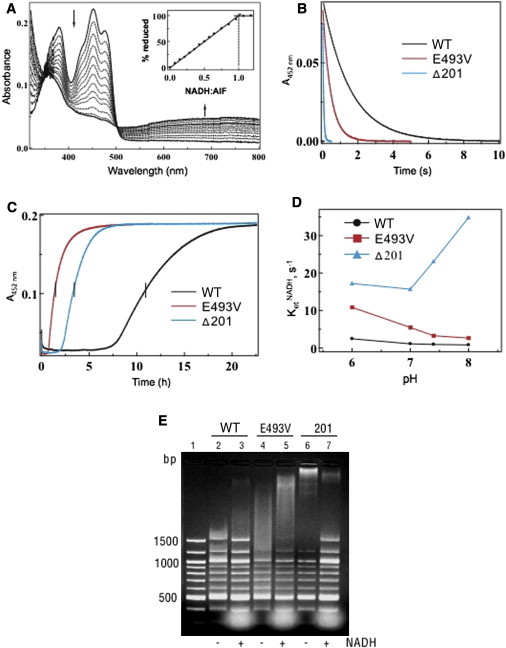
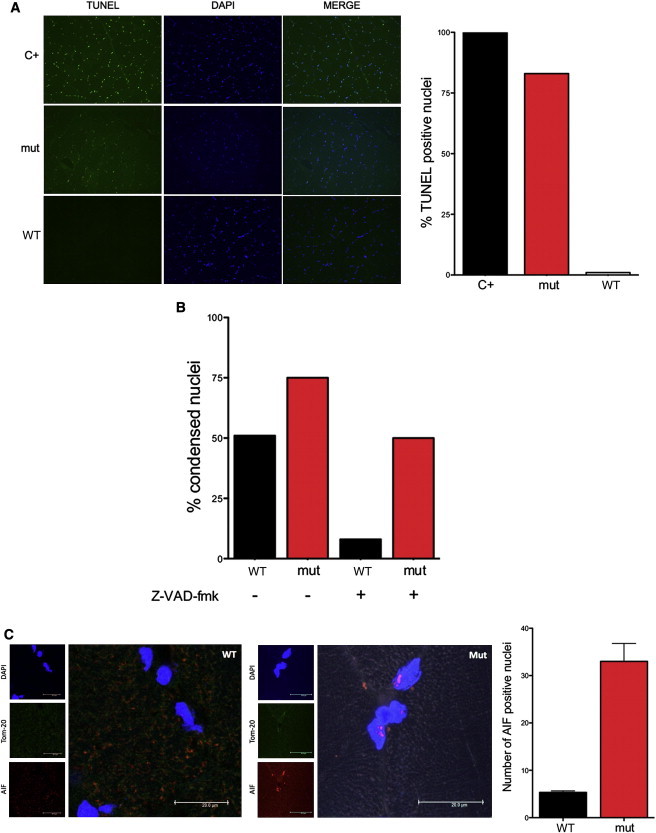
Cowchock syndrome (CMTX4) is a slowly progressive X-linked recessive disorder with axonal neuropathy, deafness, and cognitive impairment. The disease locus was previously mapped to an 11 cM region at chromosome X: q24-q26. Exome sequencing of an affected individual from the originally described family identified a missense change c.1478A>T (p.Glu493Val) in AIFM1, the gene encoding apoptosis-inducing factor (AIF) mitochondrion-associated 1. The change is at a highly conserved residue and cosegregated with the phenotype in the family. AIF is an FAD-dependent NADH oxidase that is imported into mitochondria. With apoptotic insults, a N-terminal transmembrane linker is cleaved off, producing a soluble fragment that is released into the cytosol and then transported into the nucleus, where it triggers caspase-independent apoptosis. Another AIFM1 mutation that predicts p.Arg201del has recently been associated with severe mitochondrial encephalomyopathy in two infants by impairing oxidative phosphorylation. The c.1478A>T (p.Glu493Val) mutation found in the family reported here alters the redox properties of the AIF protein and results in increased cell death via apoptosis, without affecting the activity of the respiratory chain complexes. Our findings expand the spectrum of AIF-related disease and provide insight into the effects of AIFM1 mutations.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974;6:98–118. - PubMed
-
- Pareyson D., Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8:654–667. - PubMed
-
- Boerkoel C.F., Takashima H., Garcia C.A., Olney R.K., Johnson J., Berry K., Russo P., Kennedy S., Teebi A.S., Scavina M. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann. Neurol. 2002;51:190–201. - PubMed
-
- Bergoffen J., Scherer S.S., Wang S., Scott M.O., Bone L.J., Paul D.L., Chen K., Lensch M.W., Chance P.F., Fischbeck K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. - PubMed
-
- Kim H.J., Sohn K.M., Shy M.E., Krajewski K.M., Hwang M., Park J.H., Jang S.Y., Won H.H., Choi B.O., Hong S.H. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5) Am. J. Hum. Genet. 2007;81:552–558. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
