

Oxidative stress and pulmonary fibrosis
- PMID: 23219955
- PMCID: PMC3639303
- DOI: 10.1016/j.bbadis.2012.11.021
Oxidative stress and pulmonary fibrosis
Abstract
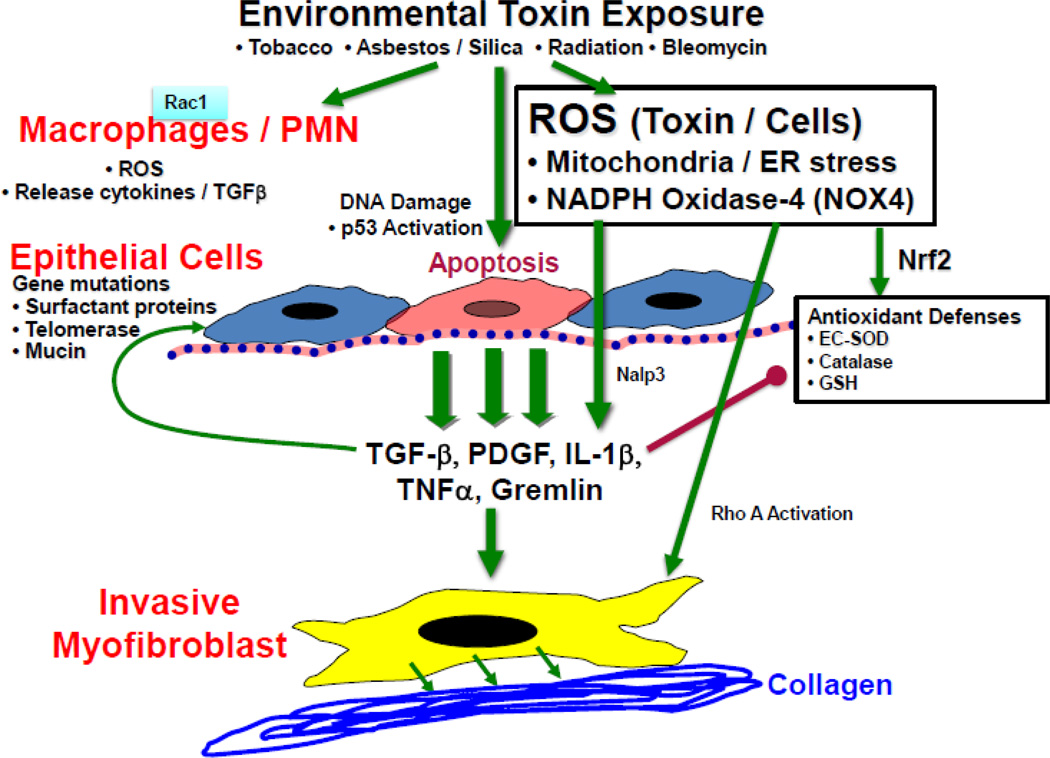
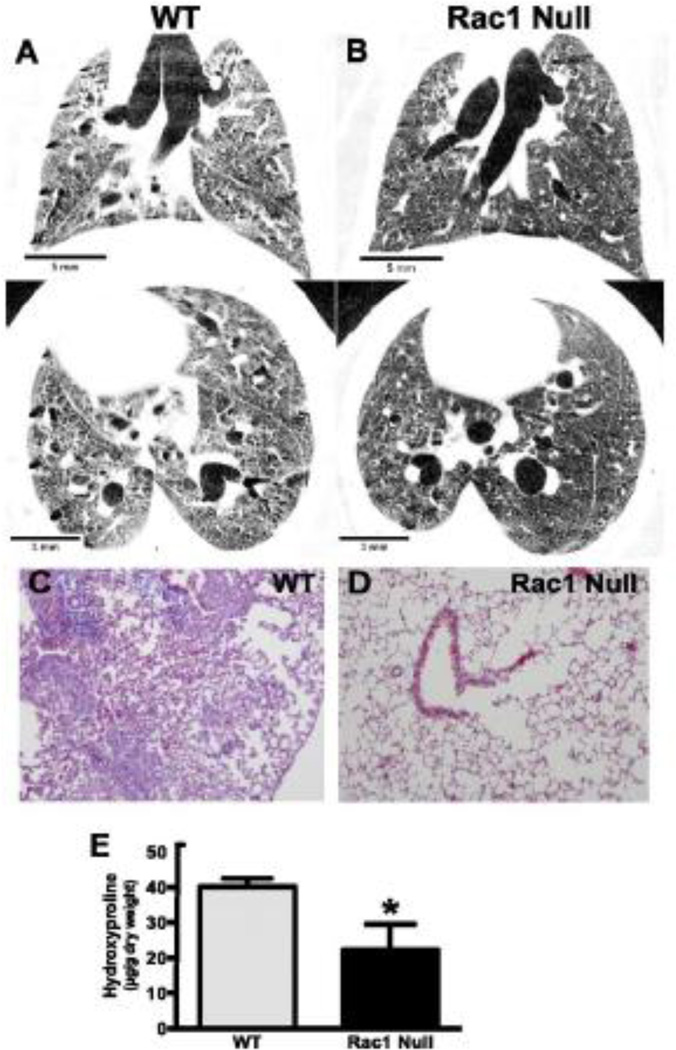
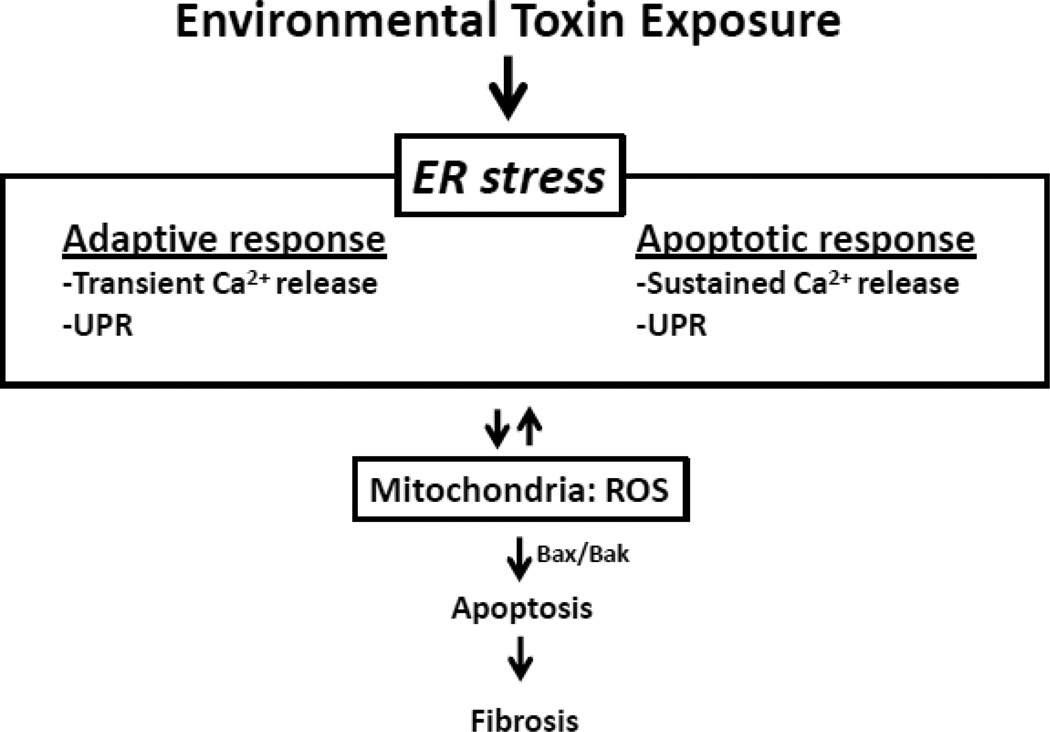
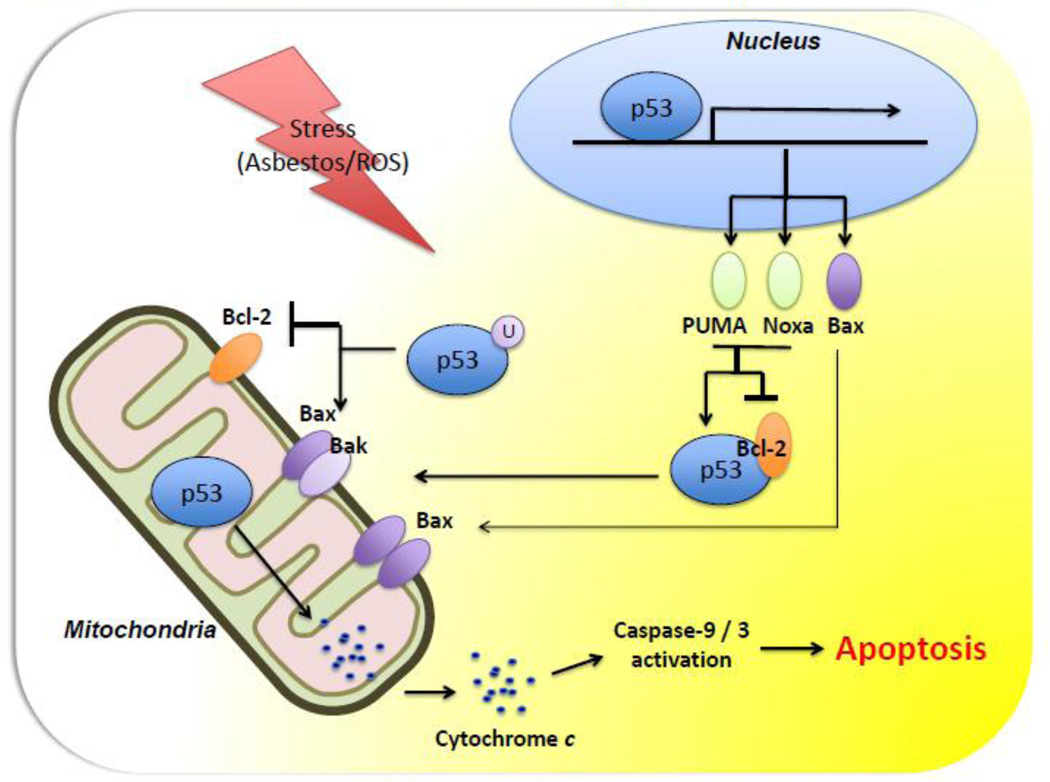
Oxidative stress is implicated as an important molecular mechanism underlying fibrosis in a variety of organs, including the lungs. However, the causal role of reactive oxygen species (ROS) released from environmental exposures and inflammatory/interstitial cells in mediating fibrosis as well as how best to target an imbalance in ROS production in patients with fibrosis is not firmly established. We focus on the role of ROS in pulmonary fibrosis and, where possible, highlight overlapping molecular pathways in other organs. The key origins of oxidative stress in pulmonary fibrosis (e.g. environmental toxins, mitochondria/NADPH oxidase of inflammatory and lung target cells, and depletion of antioxidant defenses) are reviewed. The role of alveolar epithelial cell (AEC) apoptosis by mitochondria- and p53-regulated death pathways is examined. We emphasize an emerging role for the endoplasmic reticulum (ER) in pulmonary fibrosis. After briefly summarizing how ROS trigger a DNA damage response, we concentrate on recent studies implicating a role for mitochondrial DNA (mtDNA) damage and repair mechanisms focusing on 8-oxoguanine DNA glycosylase (Ogg1) as well as crosstalk between ROS production, mtDNA damage, p53, Ogg1, and mitochondrial aconitase (ACO2). Finally, the association between ROS and TGF-β1-induced fibrosis is discussed. Novel insights into the molecular basis of ROS-induced pulmonary diseases and, in particular, lung epithelial cell death may promote the development of unique therapeutic targets for managing pulmonary fibrosis as well as fibrosis in other organs and tumors, and in aging; diseases for which effective management is lacking. This article is part of a Special Issue entitled: Fibrosis: Translation of basic research to human disease.
Published by Elsevier B.V.
Figures
References
-
- Faner R, Rojas M, Macnee W, Agusti A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012;186:306–313. - PubMed
-
- Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir. Crit. Care. Med. 2003;167:1600–1619. - PubMed
-
- Jones DP. Extracellular redox state: refining the definition of oxidative stress in aging. Rejuvenation Res. 2006;9:169–181. - PubMed
-
- Sanchez-Valle V, Chavez-Tapia NC, Uribe M, Mendez-Sanchez N. Role of Oxidative Stress and Molecular Changes in Liver Fibrosis: A Review. Curr. Med. Chem. 2012 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
