

ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses
- PMID: 23221566
- PMCID: PMC3571479
- DOI: 10.1128/JVI.02097-12
ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses
Abstract
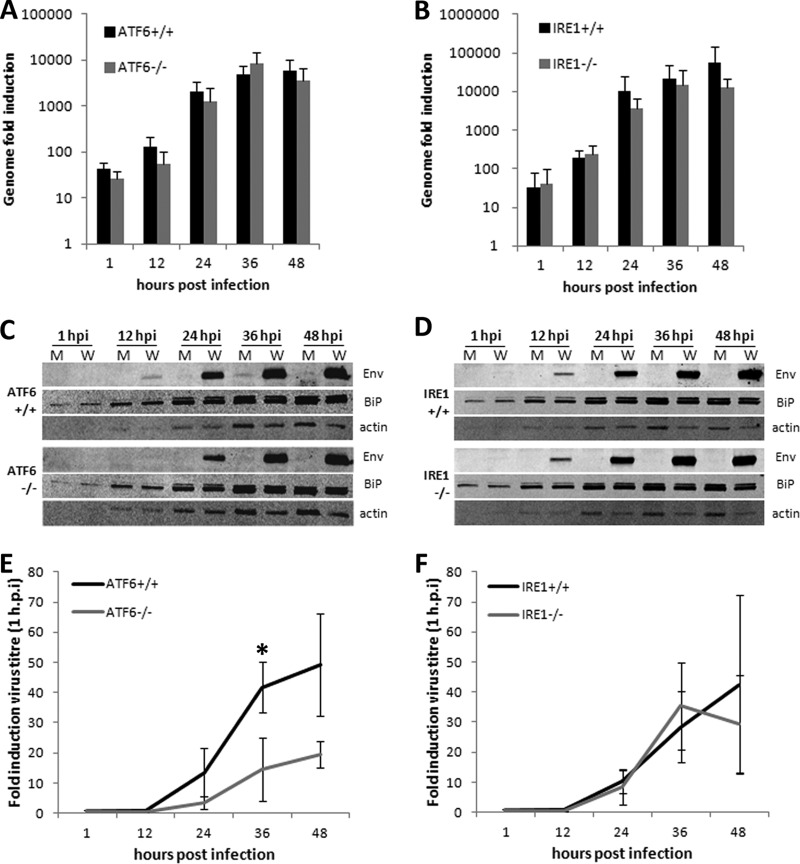
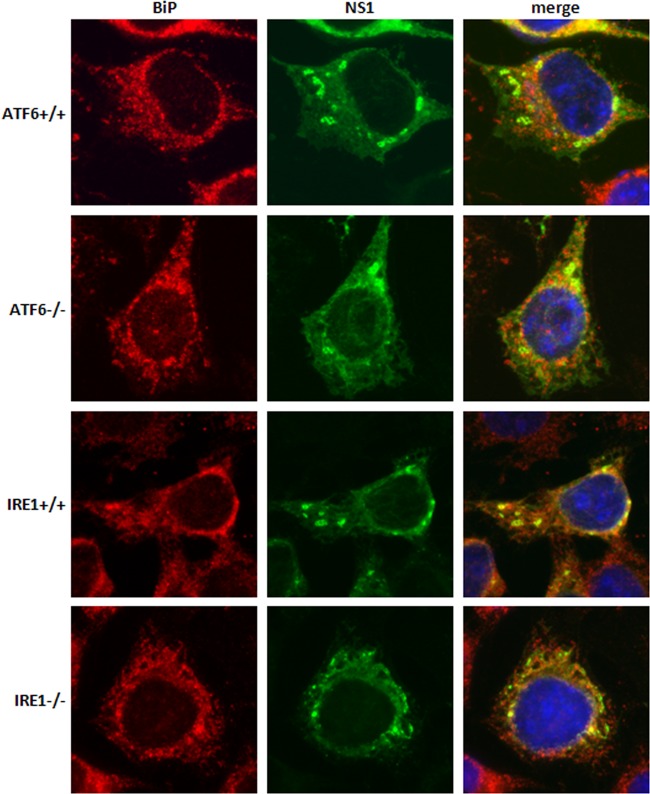
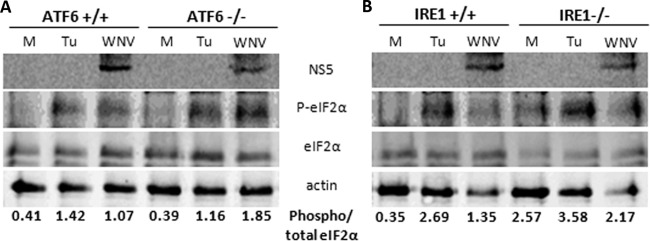
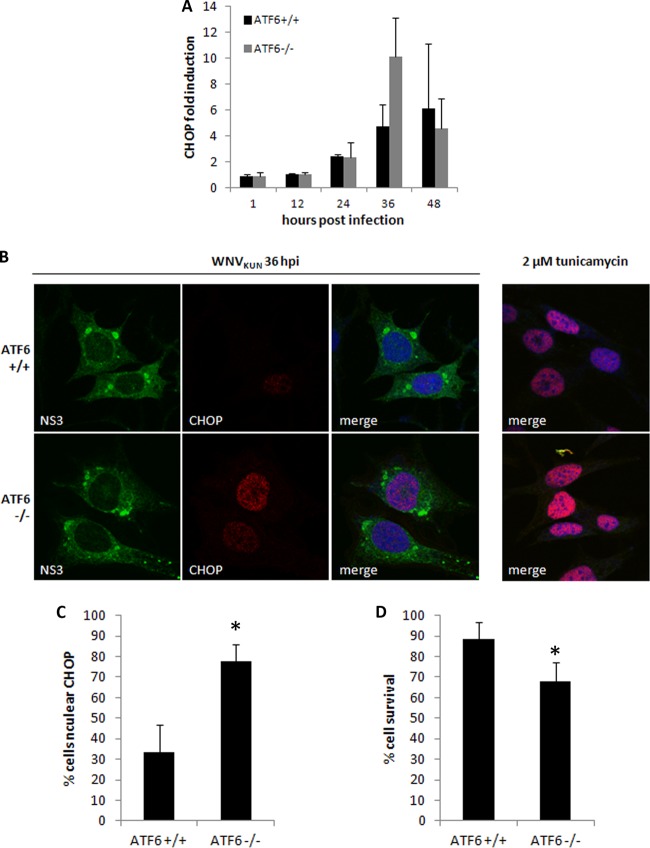
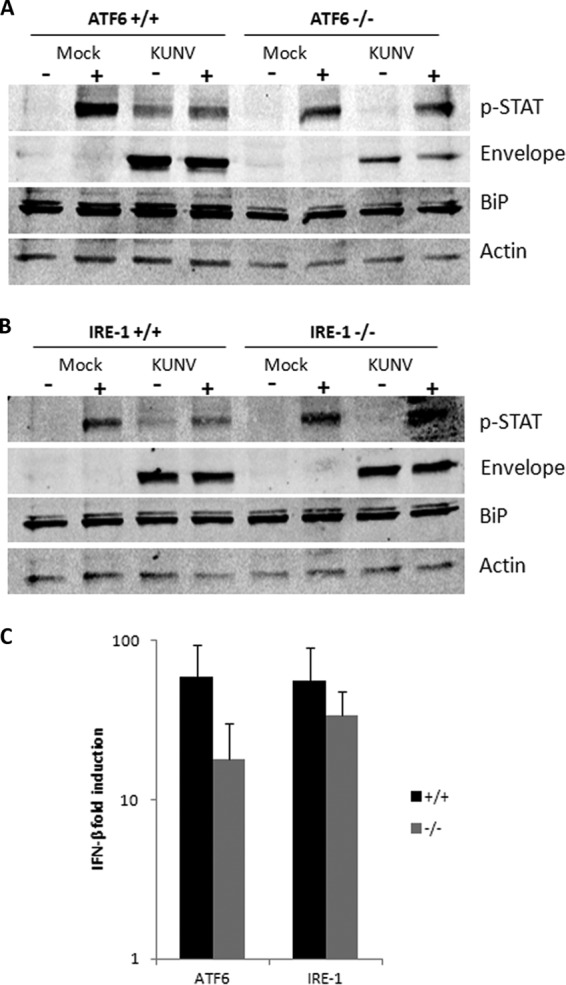
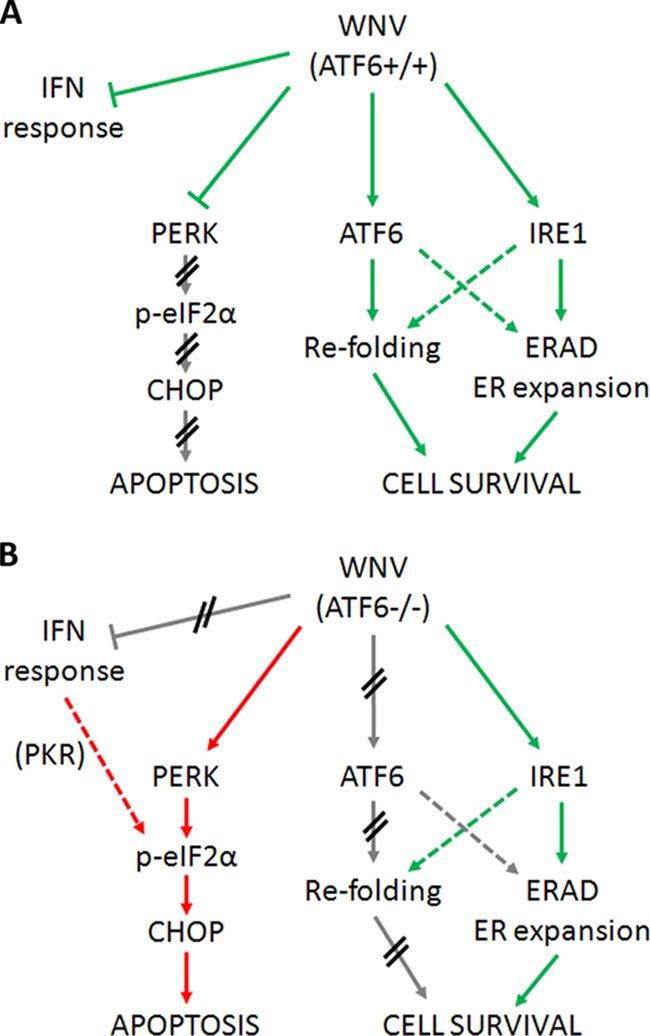
West Nile virus strain Kunjin (WNV(KUN)) is an enveloped, positive-sense RNA virus within the virus family Flaviviridae. Many flaviviruses have been shown to manipulate multiple signaling pathways, including autophagic, innate immune, and stress responses, in order to benefit replication. In particular, we have demonstrated that WNV(KUN) regulates the unfolded protein response (UPR), skewing the downstream effectors toward chaperone expression and Xbp-1 activation while preventing PERK-mediated translation attenuation and C/EBP homologous protein (CHOP) upregulation. WNV(KUN)-regulated UPR signaling can then be hijacked in order to affect type I interferon (IFN) responses, preventing IFN-mediated STAT1 phosphorylation and nuclear translocation. To extend our previous observations, we aimed to investigate the contribution of ATF6- and IRE1-mediated signaling during WNV(KUN) replication and how the two sensors contribute to the inhibition of IFN signaling. ATF6-deficient cells infected with WNV(KUN) showed decreased protein and virion production. These cells also demonstrated increased eIF2α phosphorylation and CHOP transcription, absent in infected matched control cells. Thus, we propose that in the absence of ATF6, WNV(KUN) is incapable of manipulating the PERK-mediated response to infection. In contrast, infection of IRE1(-/-) knockout cells showed no discernible differences compared to IRE1(+/+) cells. However, both ATF6 and IRE1 were required for WNV(KUN)-induced inhibition of STAT1 phosphorylation. We suggest that the combination of abhorrent UPR signaling, promotion of cell death, and increased innate immune responses contributes to the replication defects in ATF6-deficient cells, thus demonstrating the dual importance of ATF6 in maintaining cell viability and modulating immune responses during WNV(KUN) infection.
Figures
References
-
- Berridge MJ. 2002. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32:235–249 - PubMed
-
- Bernales S, Papa FR, Walter P. 2006. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 22:487–508 - PubMed
-
- Liu CY, Schroder M, Kaufman RJ. 2000. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 275:24881–24885 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
