

Genomic variation landscape of the human gut microbiome
- PMID: 23222524
- PMCID: PMC3536929
- DOI: 10.1038/nature11711
Genomic variation landscape of the human gut microbiome
Abstract
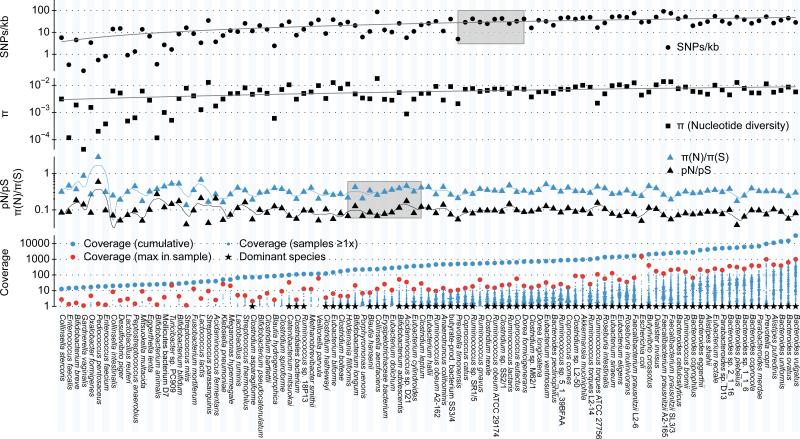
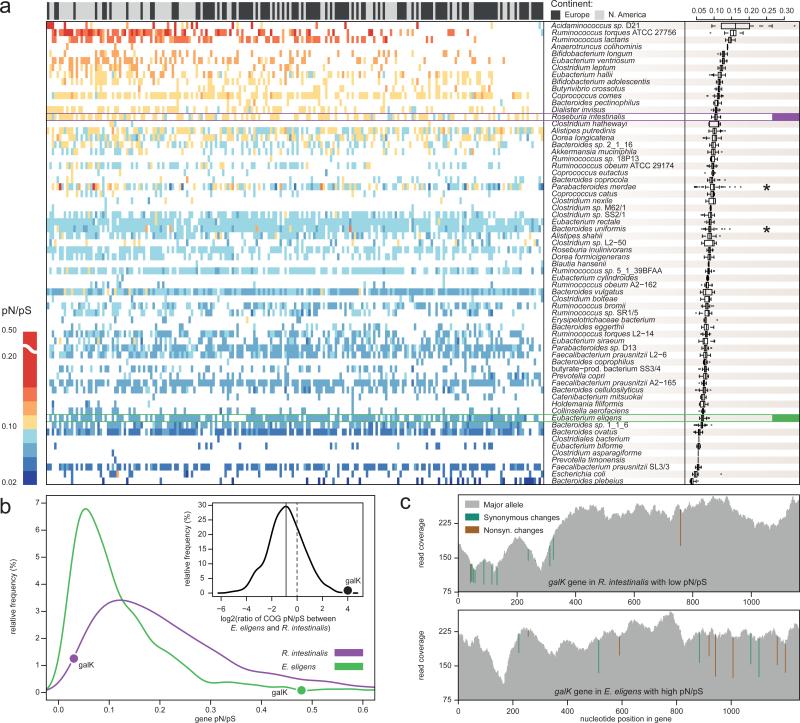
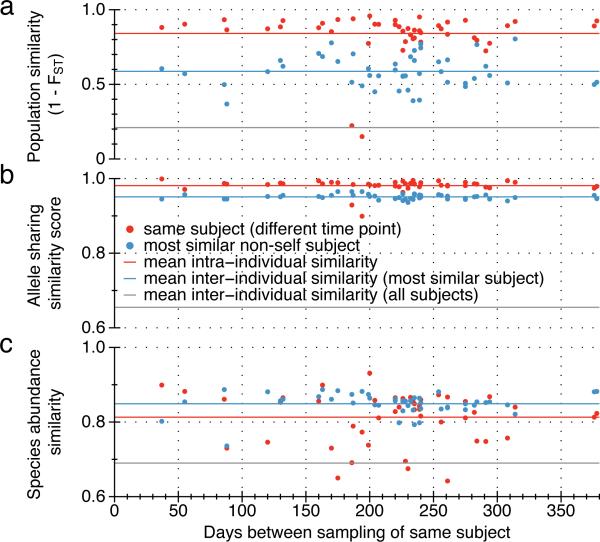
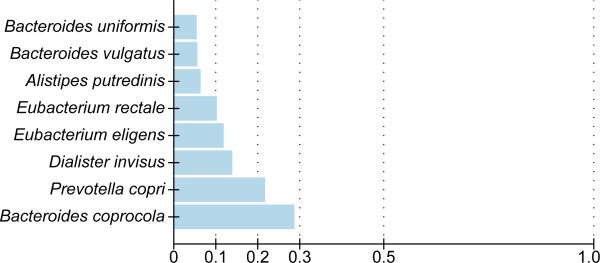
Whereas large-scale efforts have rapidly advanced the understanding and practical impact of human genomic variation, the practical impact of variation is largely unexplored in the human microbiome. We therefore developed a framework for metagenomic variation analysis and applied it to 252 faecal metagenomes of 207 individuals from Europe and North America. Using 7.4 billion reads aligned to 101 reference species, we detected 10.3 million single nucleotide polymorphisms (SNPs), 107,991 short insertions/deletions, and 1,051 structural variants. The average ratio of non-synonymous to synonymous polymorphism rates of 0.11 was more variable between gut microbial species than across human hosts. Subjects sampled at varying time intervals exhibited individuality and temporal stability of SNP variation patterns, despite considerable composition changes of their gut microbiota. This indicates that individual-specific strains are not easily replaced and that an individual might have a unique metagenomic genotype, which may be exploitable for personalized diet or drug intake.
Figures
Comment in
-
Metagenomics: Personalized gut microbiome variants.Nat Rev Genet. 2013 Feb;14(2):80. doi: 10.1038/nrg3408. Epub 2012 Dec 18. Nat Rev Genet. 2013. PMID: 23247403 No abstract available.
References
-
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. - PubMed
-
- Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi:10.1146/annurev.nutr.22.011602.092259. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
