

Good codons, bad transcript: large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme
- PMID: 23223712
- PMCID: PMC3563975
- DOI: 10.1093/molbev/mss273
Good codons, bad transcript: large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme
Abstract
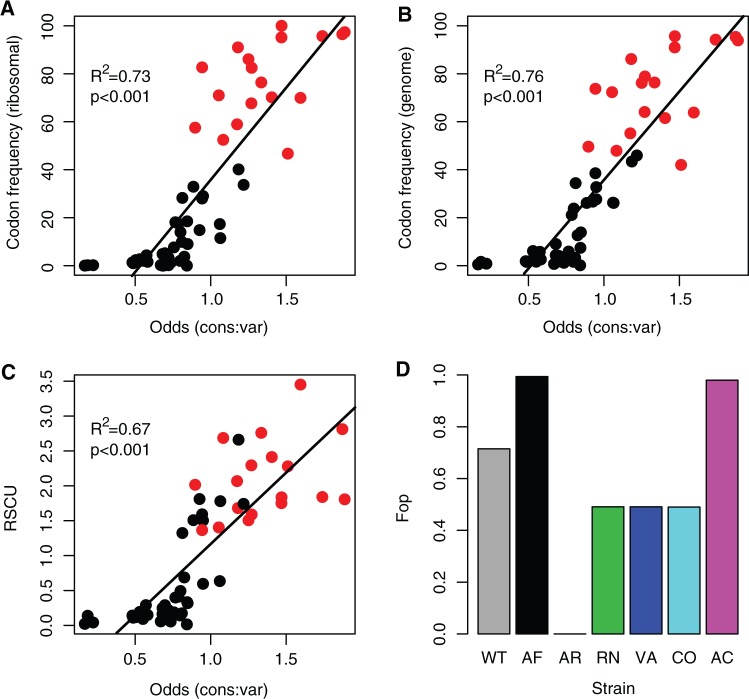
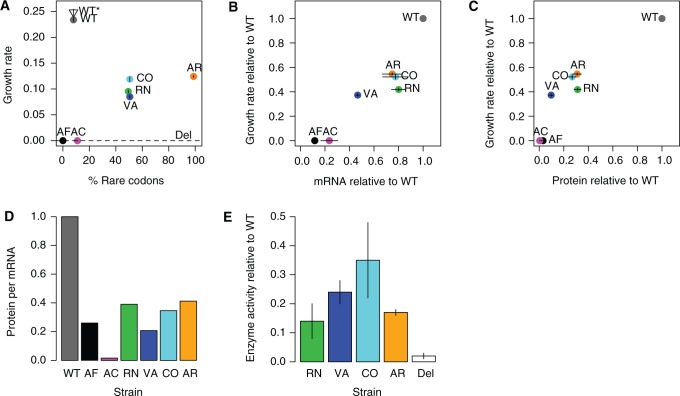
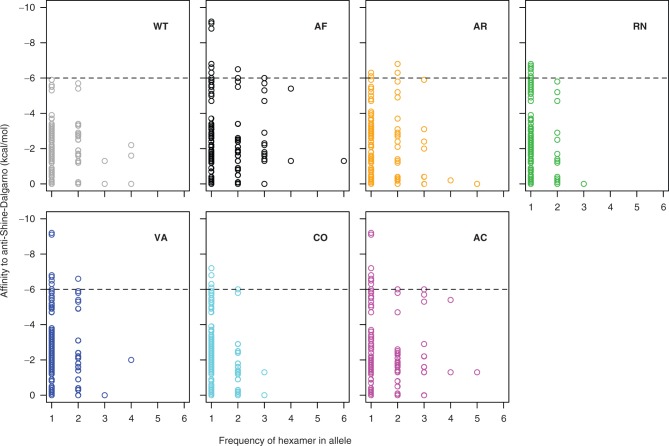
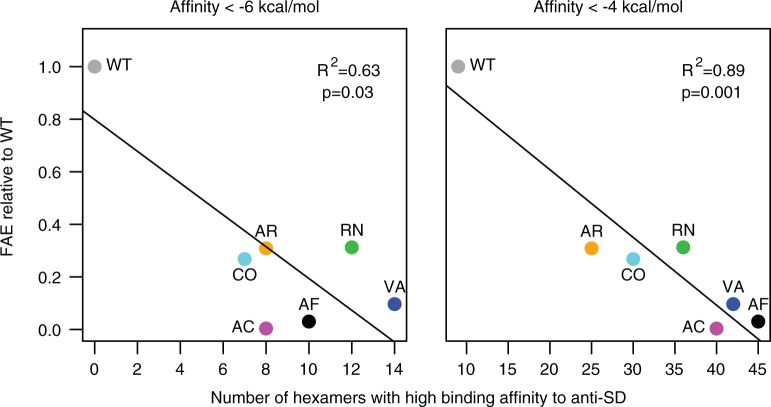
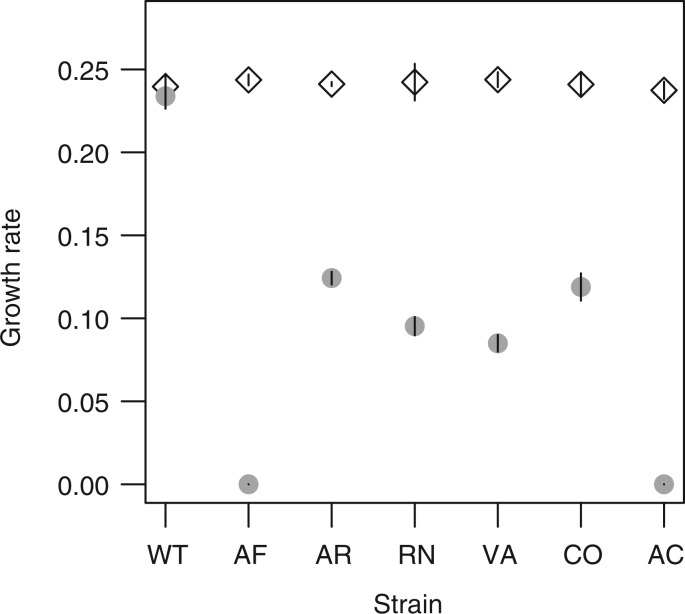
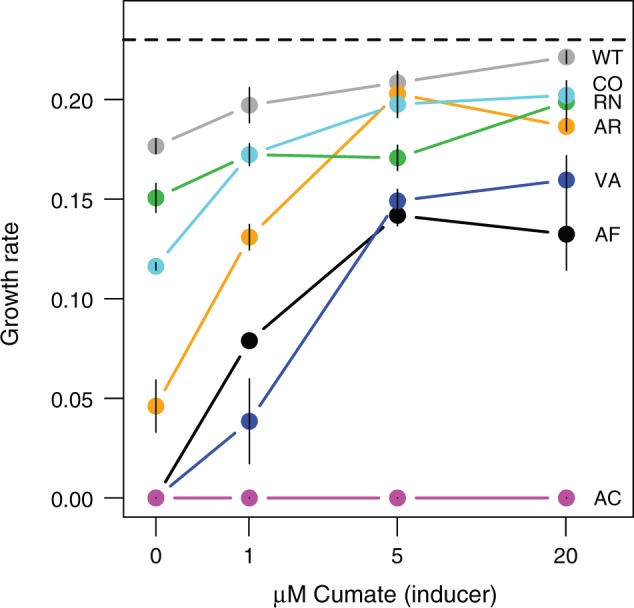
Biased codon usage in protein-coding genes is pervasive, whereby amino acids are largely encoded by a specific subset of possible codons. Within individual genes, codon bias is stronger at evolutionarily conserved residues, favoring codons recognized by abundant tRNAs. Although this observation suggests an overall pattern of selection for translation speed and/or accuracy, other work indicates that transcript structure or binding motifs drive codon usage. However, our understanding of codon bias evolution is constrained by limited experimental data on the fitness effects of altering codons in functional genes. To bridge this gap, we generated synonymous variants of a key enzyme-coding gene in Methylobacterium extorquens. We found that mutant gene expression, enzyme production, enzyme activity, and fitness were all significantly lower than wild-type. Surprisingly, encoding the gene using only rare codons decreased fitness by 40%, whereas an allele coded entirely by frequent codons decreased fitness by more than 90%. Increasing gene expression restored mutant fitness to varying degrees, demonstrating that the fitness disadvantage of synonymous mutants arose from a lack of beneficial protein rather than costs of protein production. Protein production was negatively correlated with the frequency of motifs with high affinity for the anti-Shine-Dalgarno sequence, suggesting ribosome pausing as the dominant cause of low mutant fitness. Together, our data support the idea that, although a particular set of codons are favored on average across a genome, in an individual gene selection can either act for or against codons depending on their local context.
Figures
References
-
- Amoros-Moya D, Bedhomme S, Hermann M, Bravo IG. Evolution in regulatory regions rapidly compensates the cost of nonoptimal codon usage. Mol Biol Evol. 2010;27:2141–2151. - PubMed
-
- Cannarozzi G, Schraudolph NN, Faty M, Rohr von P, Friberg MT, Roth AC, Gonnet P, Gonnet G, Barral Y. A role for codon order in translation dynamics. Cell. 2010;141:355–367. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
