

Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease
- PMID: 23224319
- PMCID: PMC3623032
- DOI: 10.1007/s00401-012-1062-9
Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease
Abstract
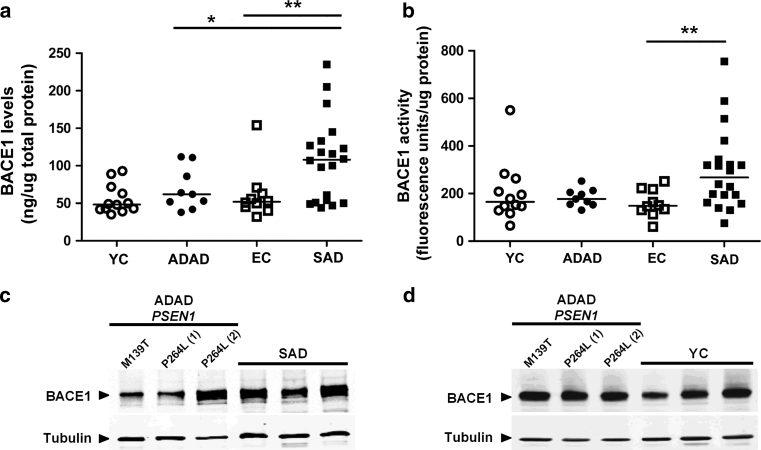
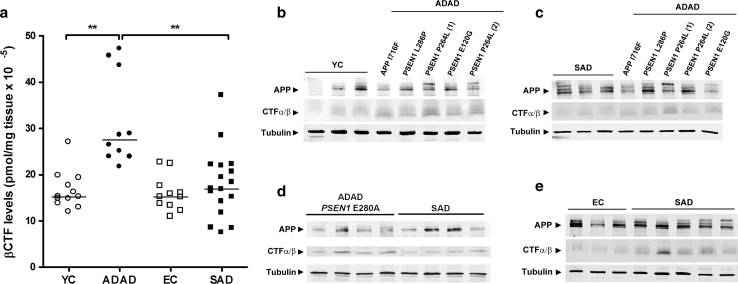
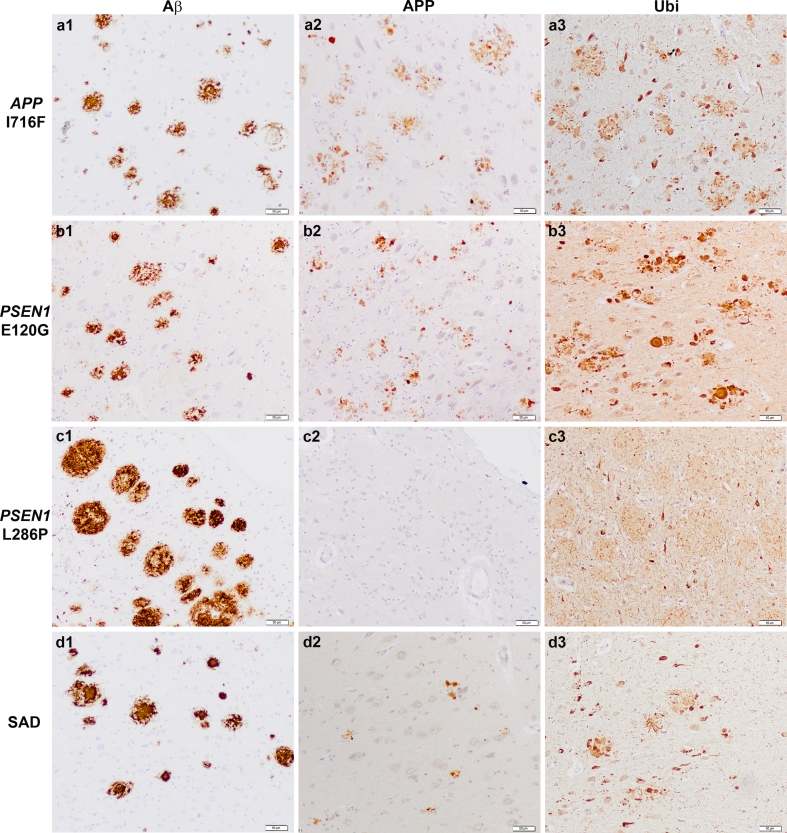
Autosomal-dominant Alzheimer disease (ADAD) is a genetic disorder caused by mutations in Amyloid Precursor Protein (APP) or Presenilin (PSEN) genes. Studies from families with ADAD have been critical to support the amyloid cascade hypothesis of Alzheimer disease (AD), the basis for the current development of amyloid-based disease-modifying therapies in sporadic AD (SAD). However, whether the pathological changes in APP processing in the CNS in ADAD are similar to those observed in SAD remains unclear. In this study, we measured β-site APP-cleaving enzyme (BACE) protein levels and activity, APP and APP C-terminal fragments in brain samples from subjects with ADAD carrying APP or PSEN1 mutations (n = 18), patients with SAD (n = 27) and age-matched controls (n = 22). We also measured sAPPβ and BACE protein levels, as well as BACE activity, in CSF from individuals carrying PSEN1 mutations (10 mutation carriers and 7 non-carrier controls), patients with SAD (n = 32) and age-matched controls (n = 11). We found that in the brain, the pattern in ADAD was characterized by an increase in APP β-C-terminal fragment (β-CTF) levels despite no changes in BACE protein levels or activity. In contrast, the pattern in SAD in the brain was mainly characterized by an increase in BACE levels and activity, with less APP β-CTF accumulation than ADAD. In the CSF, no differences were found between groups in BACE activity or expression or sAPPβ levels. Taken together, these data suggest that the physiopathological events underlying the chronic Aβ production/clearance imbalance in SAD and ADAD are different. These differences should be considered in the design of intervention trials in AD.
Figures

Similar articles
-
CNS amyloid-β, soluble APP-α and -β kinetics during BACE inhibition.J Neurosci. 2014 Jun 11;34(24):8336-46. doi: 10.1523/JNEUROSCI.0540-14.2014. J Neurosci. 2014. PMID: 24920637 Free PMC article.
-
Spectrum of γ-Secretase dysfunction as a unifying predictor of ADAD age at onset across PSEN1, PSEN2 and APP causal genes.Mol Neurodegener. 2025 Apr 26;20(1):48. doi: 10.1186/s13024-025-00832-1. Mol Neurodegener. 2025. PMID: 40281586 Free PMC article.
-
Autosomal-dominant Alzheimer's disease mutations at the same codon of amyloid precursor protein differentially alter Aβ production.J Neurochem. 2014 Jan;128(2):330-9. doi: 10.1111/jnc.12466. Epub 2013 Oct 24. J Neurochem. 2014. PMID: 24117942
-
Alzheimer's disease.Subcell Biochem. 2012;65:329-52. doi: 10.1007/978-94-007-5416-4_14. Subcell Biochem. 2012. PMID: 23225010 Review.
-
Closing in on the amyloid cascade: recent insights into the cell biology of Alzheimer's disease.Mol Neurobiol. 2000 Aug-Dec;22(1-3):81-98. doi: 10.1385/MN:22:1-3:081. Mol Neurobiol. 2000. PMID: 11414282 Review.
Cited by
-
Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology.Acta Neuropathol. 2016 Aug;132(2):257-276. doi: 10.1007/s00401-016-1577-6. Epub 2016 Apr 30. Acta Neuropathol. 2016. PMID: 27138984 Free PMC article.
-
Amyloid in the ageing brain: New frameworks and perspectives.Aging Brain. 2021 Feb 12;1:100008. doi: 10.1016/j.nbas.2021.100008. eCollection 2021. Aging Brain. 2021. PMID: 36911501 Free PMC article. No abstract available.
-
Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights From Alternative Hypotheses.Front Neurosci. 2019 May 7;13:446. doi: 10.3389/fnins.2019.00446. eCollection 2019. Front Neurosci. 2019. PMID: 31133787 Free PMC article. Review.
-
Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease.EMBO J. 2017 Nov 15;36(22):3356-3371. doi: 10.15252/embj.201796797. Epub 2017 Oct 10. EMBO J. 2017. PMID: 29018038 Free PMC article.
-
Concomitant Retinal Alterations in Neuronal Activity and TNFα Pathway Are Detectable during the Pre-Symptomatic Stage in a Mouse Model of Alzheimer's Disease.Cells. 2022 May 16;11(10):1650. doi: 10.3390/cells11101650. Cells. 2022. PMID: 35626688 Free PMC article.
References
-
- Alafuzoff I, Pikkarainen M, Arzberger T, Thal DR, Al-Sarraj S, Bell J, Bodi I, Budka H, Capetillo-Zarate E, Ferrer I, Gelpi E, Gentleman S, Giaccone G, Kavantzas N, King A, Korkolopoulou P, Kovacs GG, Meyronet D, Monoranu C, Parchi P, Patsouris E, Roggendorf W, Stadelmann C, Streichenberger N, Tagliavini F, Kretzschmar H. Inter-laboratory comparison of neuropathological assessments of beta-amyloid protein: a study of the BrainNet Europe consortium. Acta Neuropathol. 2008;115(5):533–546. doi: 10.1007/s00401-008-0358-2. - DOI - PubMed
-
- Aso E, Lomoio S, Lopez-Gonzalez I, Joda L, Carmona M, Fernandez-Yague N, Moreno J, Juves S, Pujol A, Pamplona R, Portero-Otin M, Martin V, Diaz M, Ferrer I. Amyloid generation and dysfunctional immunoproteasome activation with disease progression in animal model of familial Alzheimer’s disease. Brain Pathol. 2011 - PMC - PubMed
-
- Balasa M, Vidal-Pineiro D, Llado A, Antonell A, Bosch B, Castellanos F, Bargallo N, Bartres-Faz D, Molinuevo JL, Sanchez-Valle R. PSEN1 mutation carriers present lower cerebrospinal fluid amyloid-β42 levels than sporadic early-onset Alzheimer’s disease patients but no differences in neuronal injury biomarkers. J Alzheimers Dis. 2012;30(3):605–616. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
