

Assessment of genotype imputation performance using 1000 Genomes in African American studies
- PMID: 23226329
- PMCID: PMC3511547
- DOI: 10.1371/journal.pone.0050610
Assessment of genotype imputation performance using 1000 Genomes in African American studies
Abstract
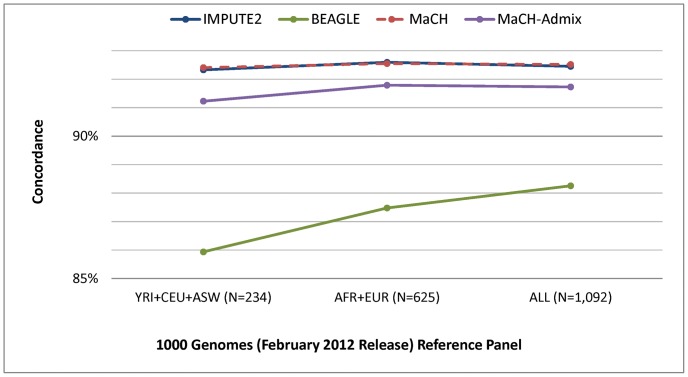
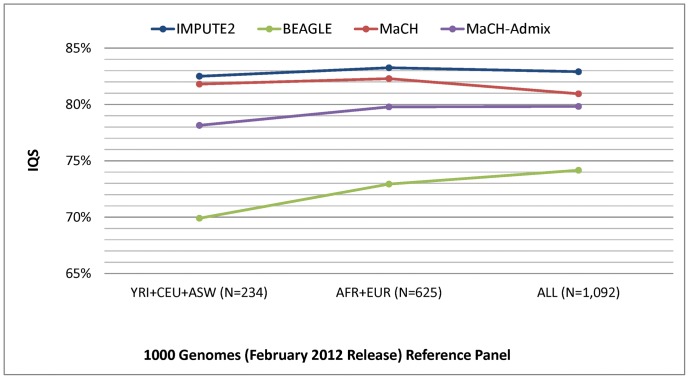
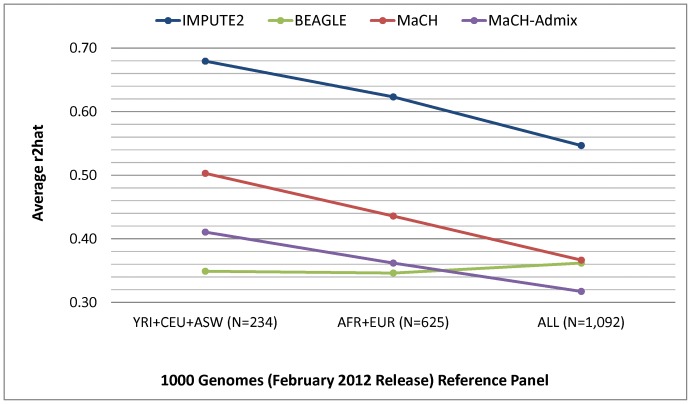
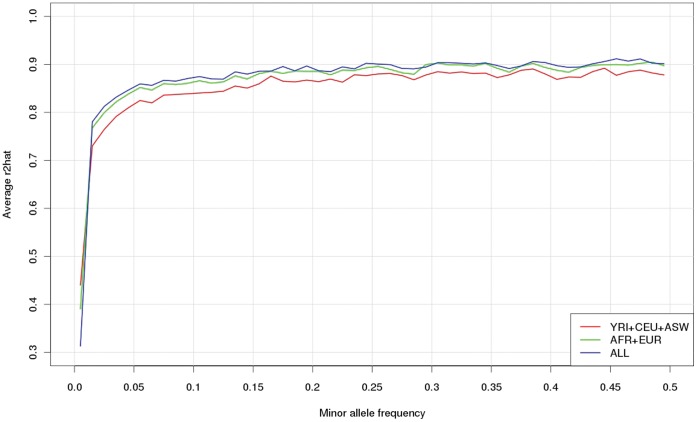
Genotype imputation, used in genome-wide association studies to expand coverage of single nucleotide polymorphisms (SNPs), has performed poorly in African Americans compared to less admixed populations. Overall, imputation has typically relied on HapMap reference haplotype panels from Africans (YRI), European Americans (CEU), and Asians (CHB/JPT). The 1000 Genomes project offers a wider range of reference populations, such as African Americans (ASW), but their imputation performance has had limited evaluation. Using 595 African Americans genotyped on Illumina's HumanHap550v3 BeadChip, we compared imputation results from four software programs (IMPUTE2, BEAGLE, MaCH, and MaCH-Admix) and three reference panels consisting of different combinations of 1000 Genomes populations (February 2012 release): (1) 3 specifically selected populations (YRI, CEU, and ASW); (2) 8 populations of diverse African (AFR) or European (AFR) descent; and (3) all 14 available populations (ALL). Based on chromosome 22, we calculated three performance metrics: (1) concordance (percentage of masked genotyped SNPs with imputed and true genotype agreement); (2) imputation quality score (IQS; concordance adjusted for chance agreement, which is particularly informative for low minor allele frequency [MAF] SNPs); and (3) average r2hat (estimated correlation between the imputed and true genotypes, for all imputed SNPs). Across the reference panels, IMPUTE2 and MaCH had the highest concordance (91%-93%), but IMPUTE2 had the highest IQS (81%-83%) and average r2hat (0.68 using YRI+ASW+CEU, 0.62 using AFR+EUR, and 0.55 using ALL). Imputation quality for most programs was reduced by the addition of more distantly related reference populations, due entirely to the introduction of low frequency SNPs (MAF≤2%) that are monomorphic in the more closely related panels. While imputation was optimized by using IMPUTE2 with reference to the ALL panel (average r2hat = 0.86 for SNPs with MAF>2%), use of the ALL panel for African American studies requires careful interpretation of the population specificity and imputation quality of low frequency SNPs.
Conflict of interest statement
Figures
Similar articles
-
Comprehensive evaluation of imputation performance in African Americans.J Hum Genet. 2012 Jul;57(7):411-21. doi: 10.1038/jhg.2012.43. Epub 2012 May 31. J Hum Genet. 2012. PMID: 22648186 Free PMC article.
-
Genotype imputation for African Americans using data from HapMap phase II versus 1000 genomes projects.Genet Epidemiol. 2012 Jul;36(5):508-16. doi: 10.1002/gepi.21647. Epub 2012 May 29. Genet Epidemiol. 2012. PMID: 22644746 Free PMC article.
-
Accuracy of genome-wide imputation of untyped markers and impacts on statistical power for association studies.BMC Genet. 2009 Jun 16;10:27. doi: 10.1186/1471-2156-10-27. BMC Genet. 2009. PMID: 19531258 Free PMC article.
-
Genotype Imputation in Genome-Wide Association Studies.Curr Protoc Hum Genet. 2019 Jun;102(1):e84. doi: 10.1002/cphg.84. Curr Protoc Hum Genet. 2019. PMID: 31216114 Review.
-
New approaches to disease mapping in admixed populations.Nat Rev Genet. 2011 Jun 28;12(8):523-8. doi: 10.1038/nrg3002. Nat Rev Genet. 2011. PMID: 21709689 Free PMC article. Review.
Cited by
-
Local and global ancestry inference and applications to genetic association analysis for admixed populations.Genet Epidemiol. 2014 Sep;38 Suppl 1(0 1):S5-S12. doi: 10.1002/gepi.21819. Genet Epidemiol. 2014. PMID: 25112189 Free PMC article.
-
Cis-Expression Quantitative Trait Loci Mapping Reveals Replicable Associations with Heroin Addiction in OPRM1.Biol Psychiatry. 2015 Oct 1;78(7):474-84. doi: 10.1016/j.biopsych.2015.01.003. Epub 2015 Jan 29. Biol Psychiatry. 2015. PMID: 25744370 Free PMC article.
-
Genetic Risk Stratification: A Paradigm Shift in Prevention of Coronary Artery Disease.JACC Basic Transl Sci. 2021 Mar 22;6(3):287-304. doi: 10.1016/j.jacbts.2020.09.004. eCollection 2021 Mar. JACC Basic Transl Sci. 2021. PMID: 33778213 Free PMC article. Review.
-
A data harmonization pipeline to leverage external controls and boost power in GWAS.Hum Mol Genet. 2022 Feb 3;31(3):481-489. doi: 10.1093/hmg/ddab261. Hum Mol Genet. 2022. PMID: 34508597 Free PMC article.
-
The Global Durum Wheat Panel (GDP): An International Platform to Identify and Exchange Beneficial Alleles.Front Plant Sci. 2020 Dec 21;11:569905. doi: 10.3389/fpls.2020.569905. eCollection 2020. Front Plant Sci. 2020. PMID: 33408724 Free PMC article.
References
-
- Marchini J, Howie B (2010) Genotype imputation for genome-wide association studies. Nat Rev Genet 11: 499–511. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
