

Metastatic pancreatic cancer is dependent on oncogenic Kras in mice
- PMID: 23226501
- PMCID: PMC3513322
- DOI: 10.1371/journal.pone.0049707
Metastatic pancreatic cancer is dependent on oncogenic Kras in mice
Abstract
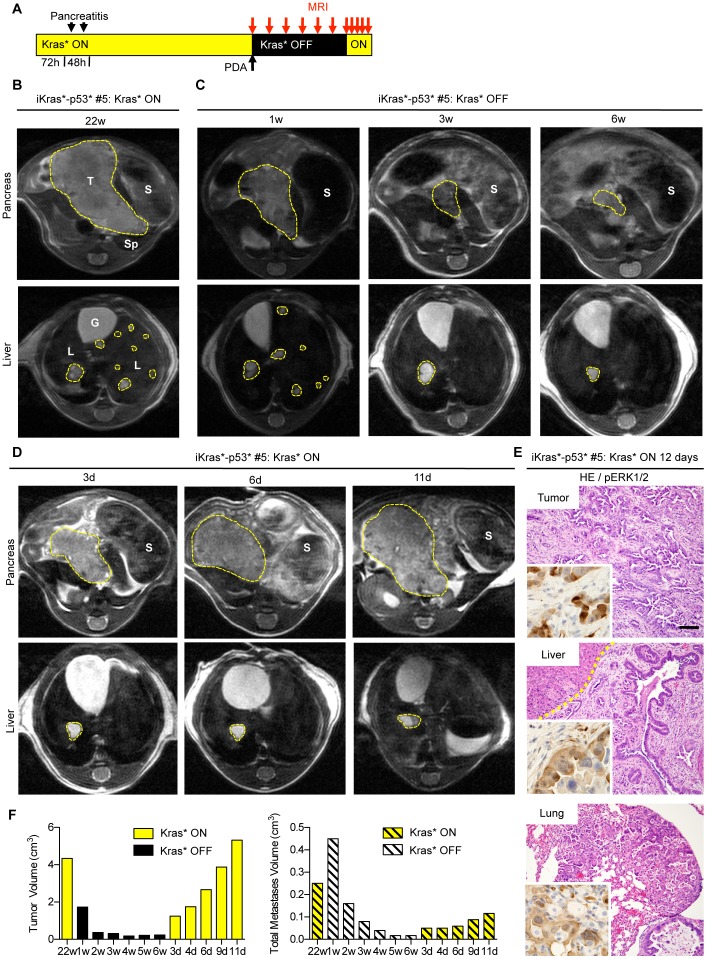
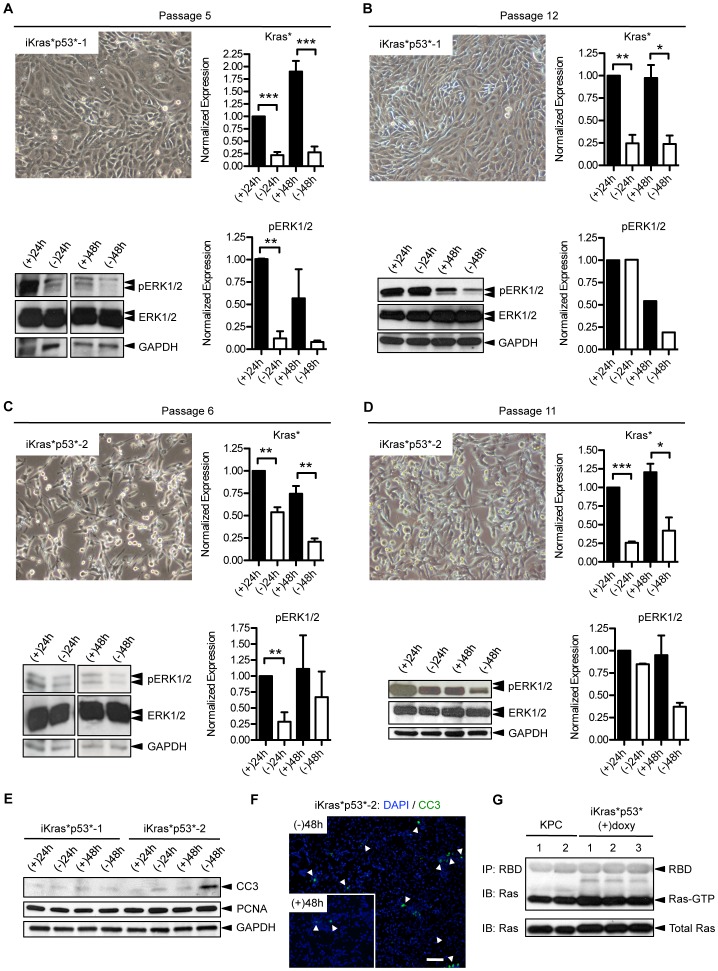
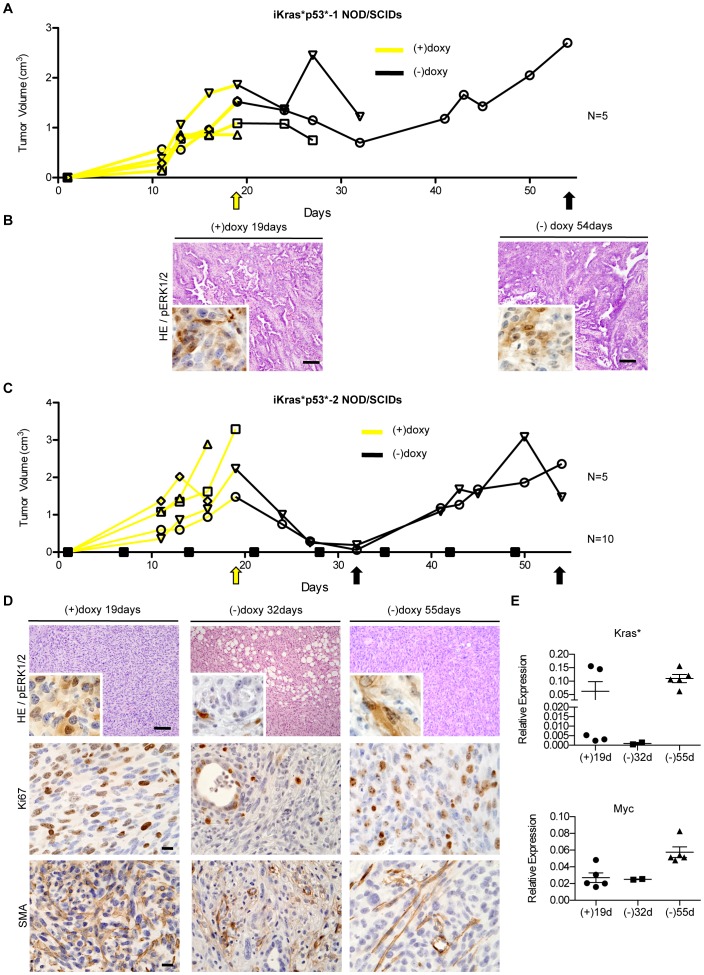
Pancreatic cancer is one of the deadliest human malignancies, and its prognosis has not improved over the past 40 years. Mouse models that spontaneously develop pancreatic adenocarcinoma and mimic the progression of the human disease are emerging as a new tool to investigate the basic biology of this disease and identify potential therapeutic targets. Here, we describe a new model of metastatic pancreatic adenocarcinoma based on pancreas-specific, inducible and reversible expression of an oncogenic form of Kras, together with pancreas-specific expression of a mutant form of the tumor suppressor p53. Using high-resolution magnetic resonance imaging to follow individual animals in longitudinal studies, we show that both primary and metastatic lesions depend on continuous Kras activity for their maintenance. However, re-activation of Kras* following prolonged inactivation leads to rapid tumor relapse, raising the concern that Kras*-resistance might eventually be acquired. Thus, our data identifies Kras* as a key oncogene in pancreatic cancer maintenance, but raises the possibility of acquired resistance should Kras inhibitors become available for use in pancreatic cancer.
Conflict of interest statement
Figures
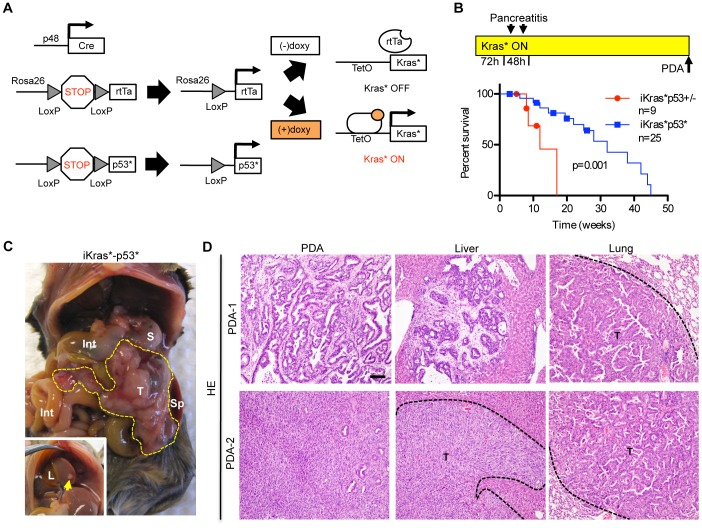
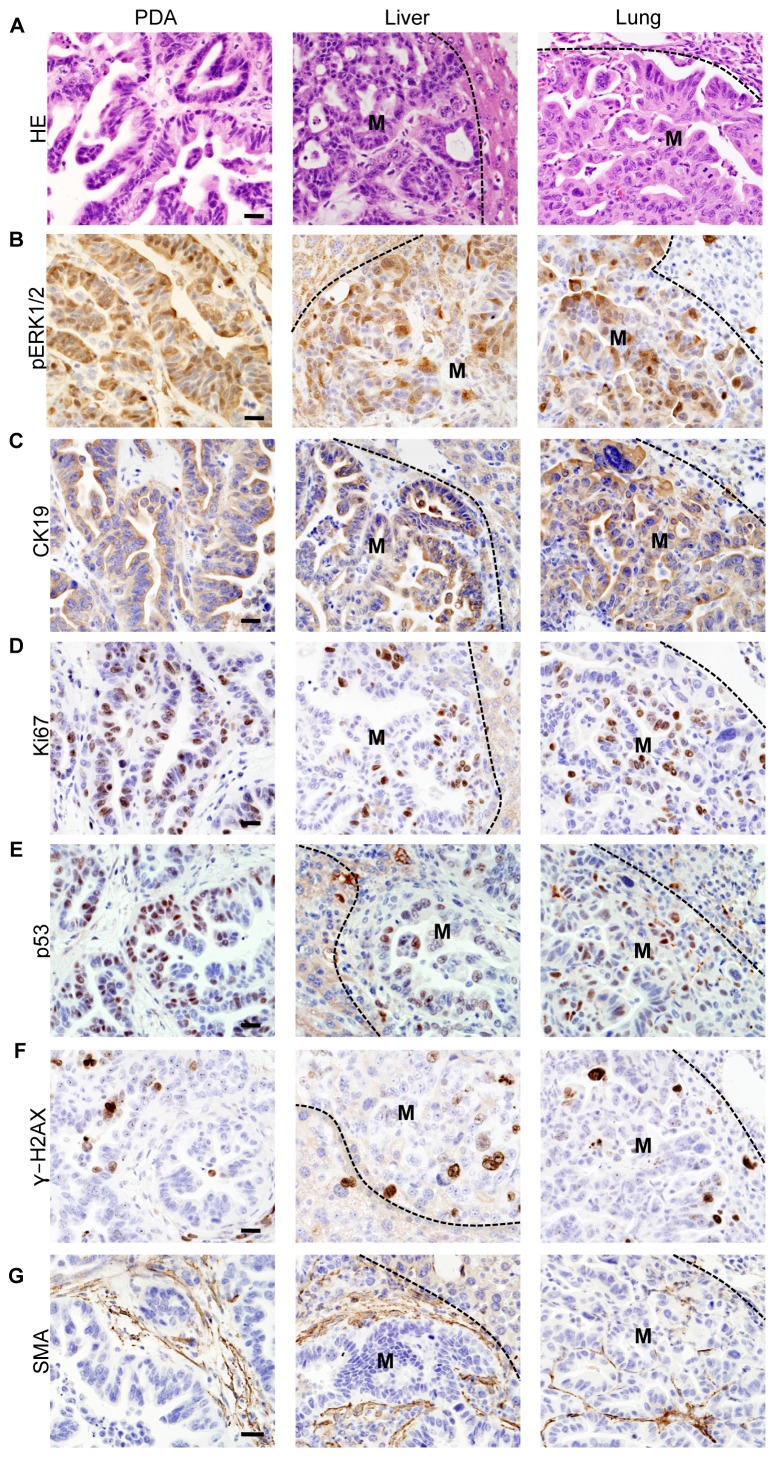
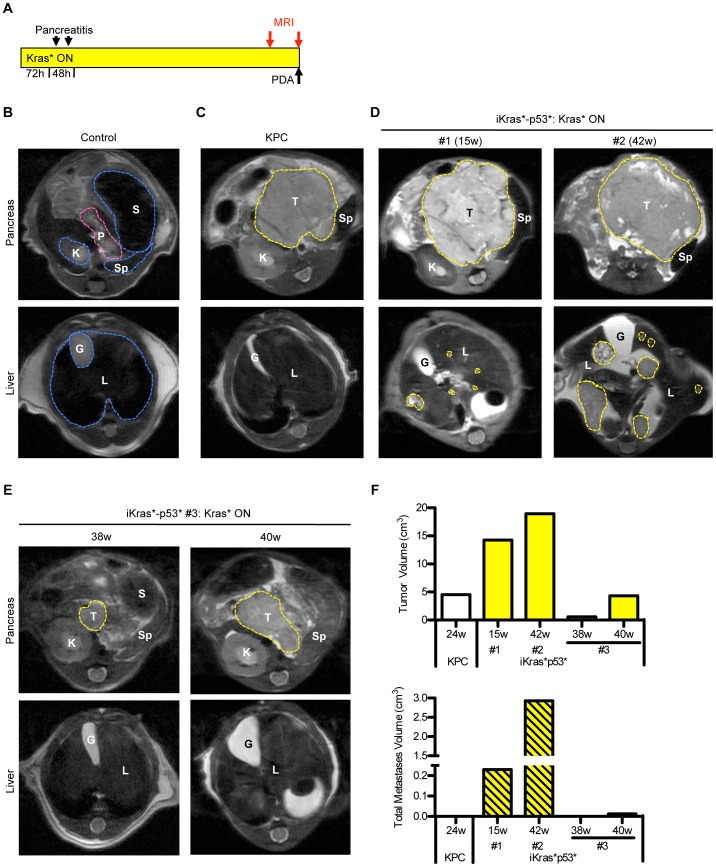
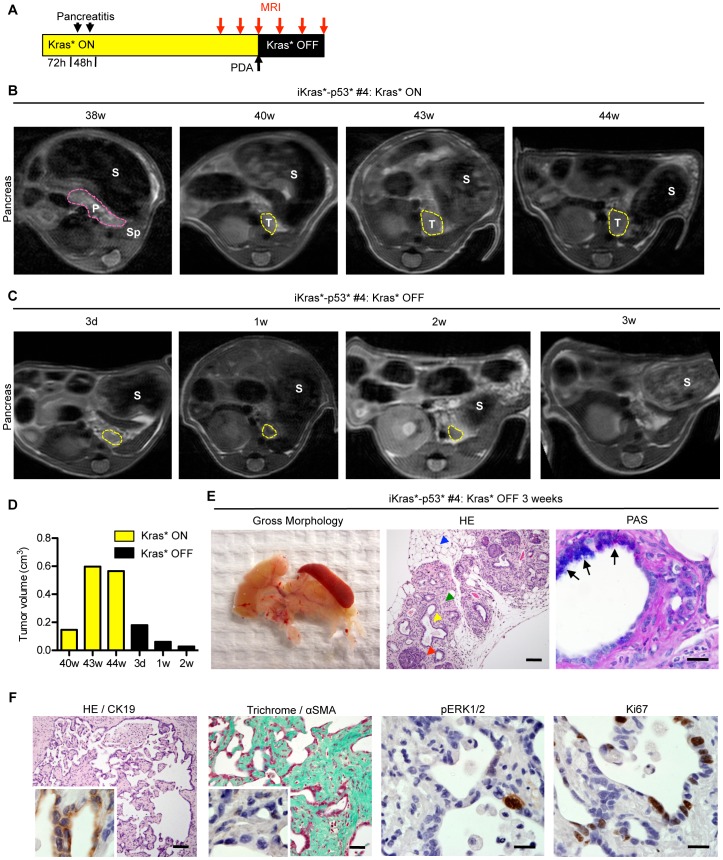
References
-
- Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, et al. (2001) Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 25: 579–586. - PubMed
-
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, et al. (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28: 622–629. - PubMed
-
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, et al. (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483. - PubMed
-
- Olive KP, Tuveson DA (2006) The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin Cancer Res 12: 5277–5287. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
