

How to infer gene networks from expression profiles, revisited
- PMID: 23226586
- PMCID: PMC3262295
- DOI: 10.1098/rsfs.2011.0053
How to infer gene networks from expression profiles, revisited
Abstract
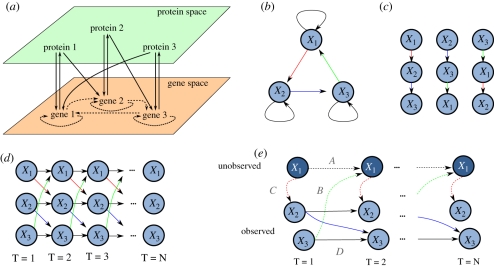
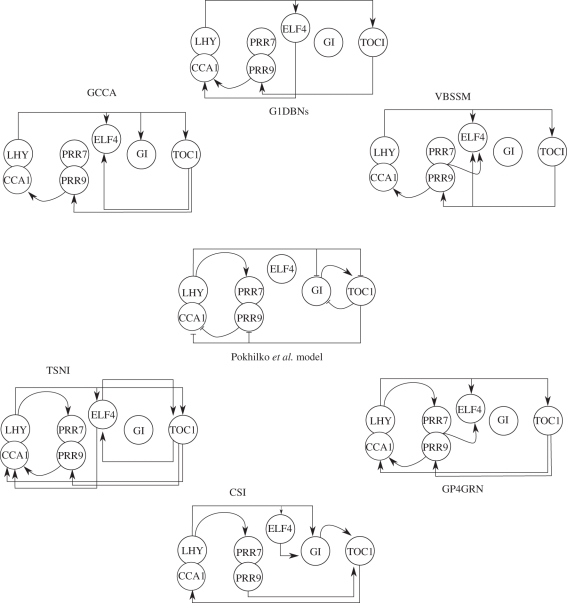
Inferring the topology of a gene-regulatory network (GRN) from genome-scale time-series measurements of transcriptional change has proved useful for disentangling complex biological processes. To address the challenges associated with this inference, a number of competing approaches have previously been used, including examples from information theory, Bayesian and dynamic Bayesian networks (DBNs), and ordinary differential equation (ODE) or stochastic differential equation. The performance of these competing approaches have previously been assessed using a variety of in silico and in vivo datasets. Here, we revisit this work by assessing the performance of more recent network inference algorithms, including a novel non-parametric learning approach based upon nonlinear dynamical systems. For larger GRNs, containing hundreds of genes, these non-parametric approaches more accurately infer network structures than do traditional approaches, but at significant computational cost. For smaller systems, DBNs are competitive with the non-parametric approaches with respect to computational time and accuracy, and both of these approaches appear to be more accurate than Granger causality-based methods and those using simple ODEs models.
Keywords: gene expression; gene-regulatory networks; inference.
Figures




References
-
- Brazhnik P., de la Fuente A., Mendes P. 2002. Gene networks: how to put the function in genomics. Trends Biotechnol. 20, 467–472 10.1016/S0167-7799(02)02053-X (doi:10.1016/S0167-7799(02)02053-X) - DOI - PubMed
-
- Park P. J. 2009. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 10.1038/nrg2641 (doi:10.1038/nrg2641) - DOI - PMC - PubMed
-
- Bulyk M. L. 2005. Discovering DNA regulatory elements with bacteria. Nat. Biotechnol. 23, 942–944 10.1038/nbt0805-942 (doi:10.1038/nbt0805-942) - DOI - PMC - PubMed
-
- Berger M. F., Bulyk M. L. 2009. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc. 4, 393–411 10.1038/nprot.2008.195 (doi:10.1038/nprot.2008.195) - DOI - PMC - PubMed
-
- Lopato S., Bazanova N., Morran S., Milligan A. S., Shirley N., Langridge P. 2006. Isolation of plant transcription factors using a modified yeast one-hybrid system. Plant Methods 2, 3. 10.1186/1746-4811-2-3 (doi:10.1186/1746-4811-2-3) - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous