

Decoding hippocampal signaling deficits after traumatic brain injury
- PMID: 23227133
- PMCID: PMC3514866
- DOI: 10.1007/s12975-011-0123-z
Decoding hippocampal signaling deficits after traumatic brain injury
Abstract
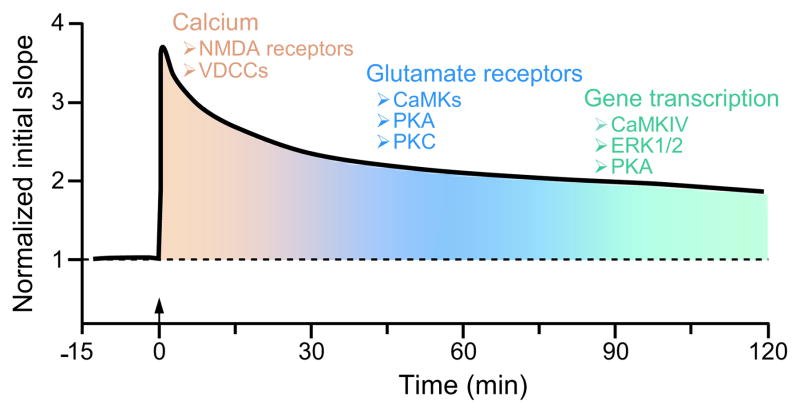
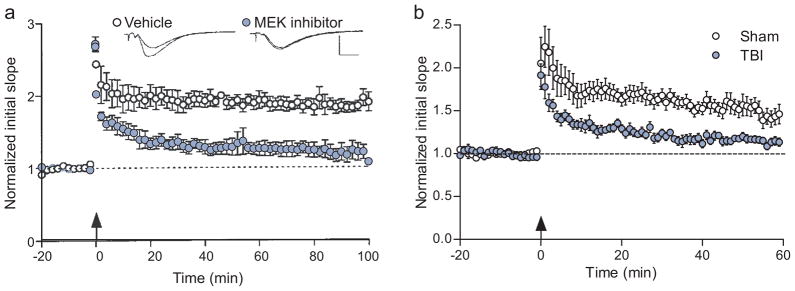
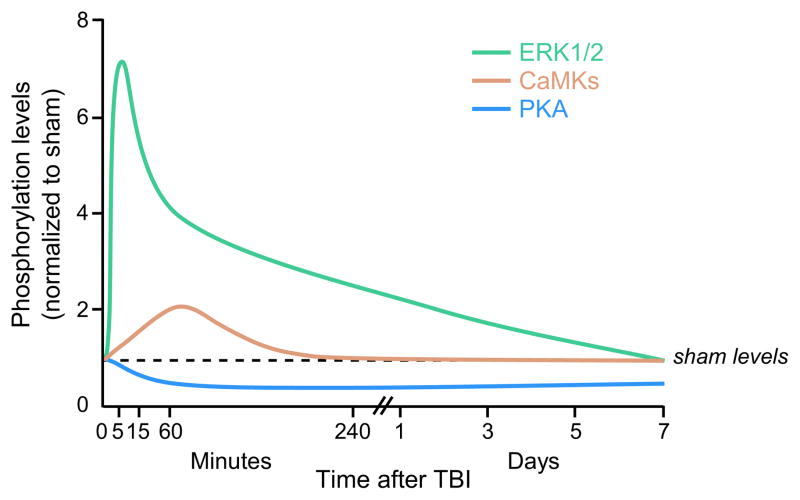
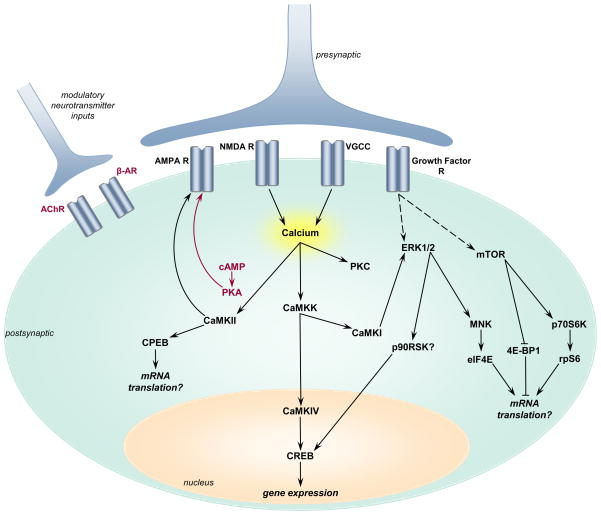
There are more than 3.17 million people coping with long-term disabilities due to traumatic brain injury (TBI) in the United States. The majority of TBI research is focused on developing acute neuroprotective treatments to prevent or minimize these long-term disabilities. Therefore, chronic TBI survivors represent a large, underserved population that could significantly benefit from a therapy that capitalizes on the endogenous recovery mechanisms occurring during the weeks to months following brain trauma. Previous studies have found that the hippocampus is highly vulnerable to brain injury, in both experimental models of TBI and during human TBI. Although often not directly mechanically injured by the head injury, in the weeks to months following TBI, the hippocampus undergoes atrophy and exhibits deficits in long-term potentiation (LTP), a persistent increase in synaptic strength that is considered to be a model of learning and memory. Decoding the chronic hippocampal LTP and cell signaling deficits after brain trauma will provide new insights into the molecular mechanisms of hippocampal-dependent learning impairments caused by TBI and facilitate the development of effective therapeutic strategies to improve hippocampal-dependent learning for chronic survivors of TBI.
Conflict of interest statement
Figures
References
-
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta, GA: 2010.
-
- Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–41. - PubMed
-
- Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59:S21–6. - PubMed
-
- Holsinger T, Steffens DC, Phillips C, Helms MJ, Havlik RJ, Breitner JC, et al. Head injury in early adulthood and the lifetime risk of depression. Arch Gen Psychiatry. 2002;59:17–22. - PubMed
-
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–66. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
