

Impact of mutations on the allosteric conformational equilibrium
- PMID: 23228330
- PMCID: PMC3557769
- DOI: 10.1016/j.jmb.2012.11.041
Impact of mutations on the allosteric conformational equilibrium
Abstract
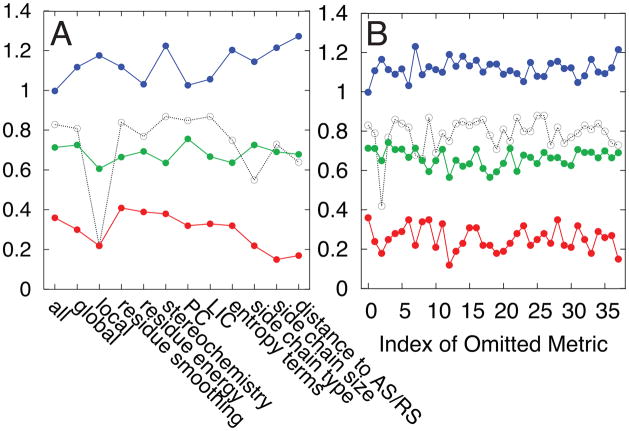
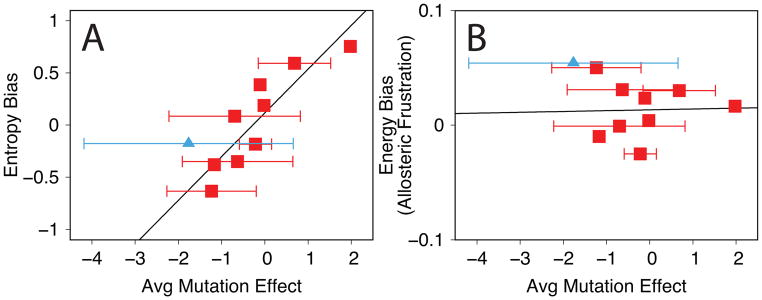
Allostery in a protein involves effector binding at an allosteric site that changes the structure and/or dynamics at a distant, functional site. In addition to the chemical equilibrium of ligand binding, allostery involves a conformational equilibrium between one protein substate that binds the effector and a second substate that less strongly binds the effector. We run molecular dynamics simulations using simple, smooth energy landscapes to sample specific ligand-induced conformational transitions, as defined by the effector-bound and effector-unbound protein structures. These simulations can be performed using our web server (http://salilab.org/allosmod/). We then develop a set of features to analyze the simulations and capture the relevant thermodynamic properties of the allosteric conformational equilibrium. These features are based on molecular mechanics energy functions, stereochemical effects, and structural/dynamic coupling between sites. Using a machine-learning algorithm on a data set of 10 proteins and 179 mutations, we predict both the magnitude and the sign of the allosteric conformational equilibrium shift by the mutation; the impact of a large identifiable fraction of the mutations can be predicted with an average unsigned error of 1k(B)T. With similar accuracy, we predict the mutation effects for an 11th protein that was omitted from the initial training and testing of the machine-learning algorithm. We also assess which calculated thermodynamic properties contribute most to the accuracy of the prediction.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures

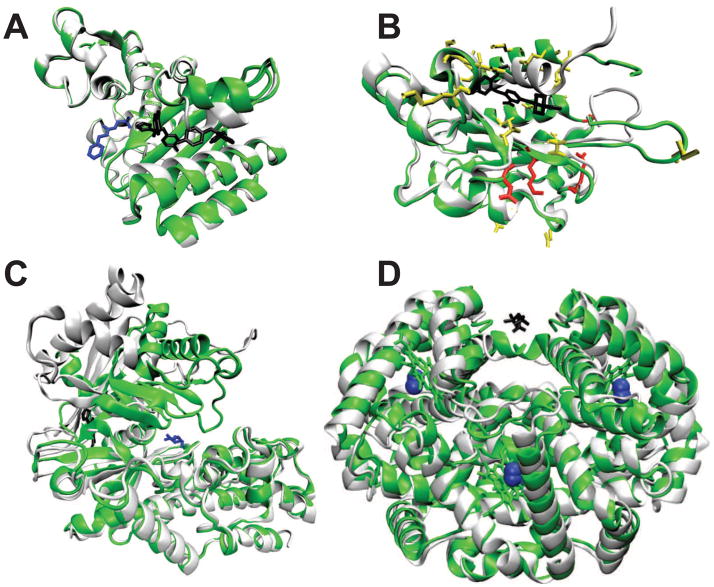
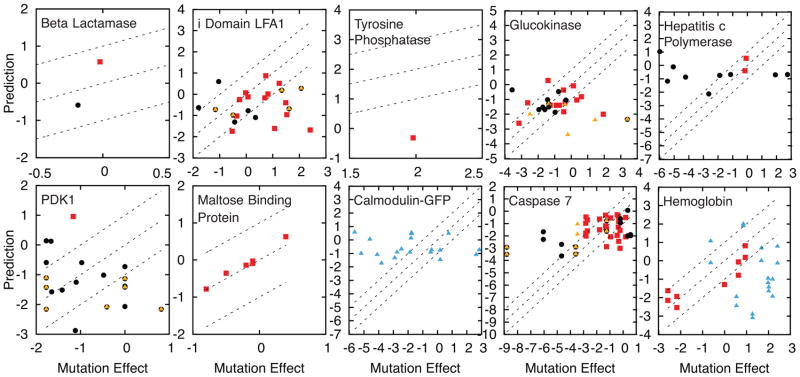
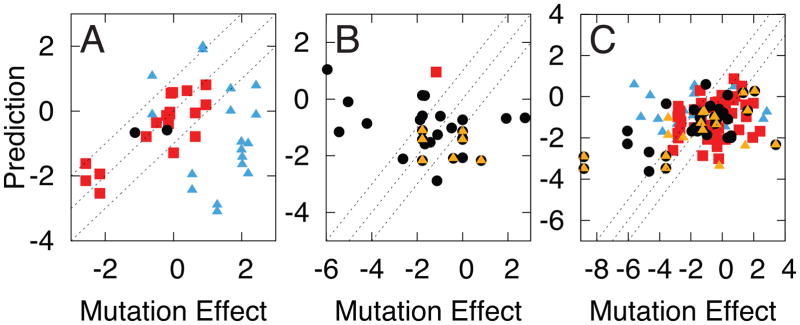
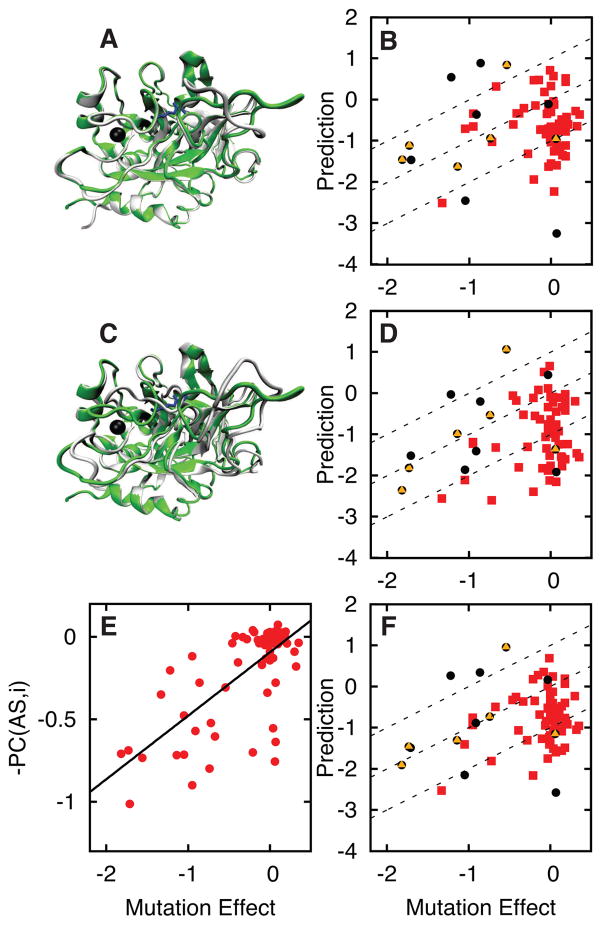
References
-
- Monod J, Wyman J, Changeux JP. On nature of allosteric transitions - a plausible model. Journal of Molecular Biology. 1965;12:88. - PubMed
-
- Koshland DE, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–368. - PubMed
-
- Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450:983–990. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
