

Genotype imputation via matrix completion
- PMID: 23233546
- PMCID: PMC3589539
- DOI: 10.1101/gr.145821.112
Genotype imputation via matrix completion
Abstract
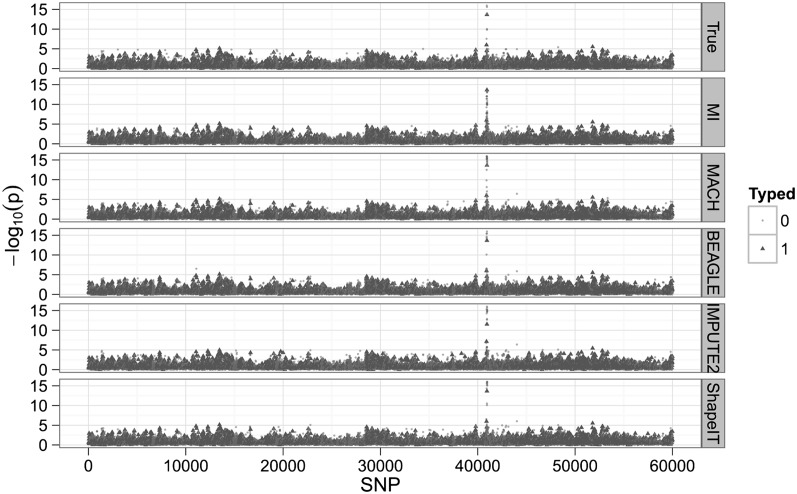
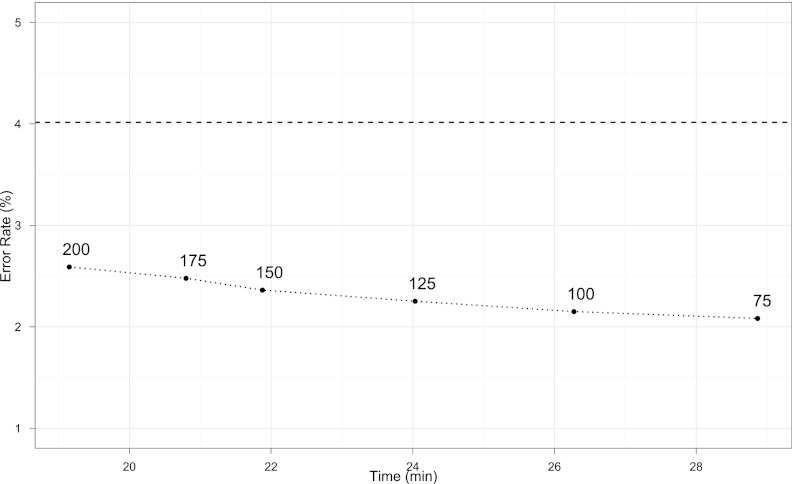
Most current genotype imputation methods are model-based and computationally intensive, taking days to impute one chromosome pair on 1000 people. We describe an efficient genotype imputation method based on matrix completion. Our matrix completion method is implemented in MATLAB and tested on real data from HapMap 3, simulated pedigree data, and simulated low-coverage sequencing data derived from the 1000 Genomes Project. Compared with leading imputation programs, the matrix completion algorithm embodied in our program MENDEL-IMPUTE achieves comparable imputation accuracy while reducing run times significantly. Implementation in a lower-level language such as Fortran or C is apt to further improve computational efficiency.
Figures
References
-
- Ayers KL, Lange K 2008. Penalized estimation of haplotype frequencies. Bioinformatics 24: 1596–1602 - PubMed
-
- Beck A, Teboulle M 2009. A fast iterative shrinkage-thresholding algorithm for linear inverse problems. SIAM J Imaging Sci 2: 183–202
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources