

Review
doi: 10.1056/NEJMra1205750.
Purinergic signaling during inflammation
Affiliations
- PMID: 23234515
- PMCID: PMC3675791
- DOI: 10.1056/NEJMra1205750
Item in Clipboard
Review
Purinergic signaling during inflammation
N Engl J Med.
.
No abstract available
Figures
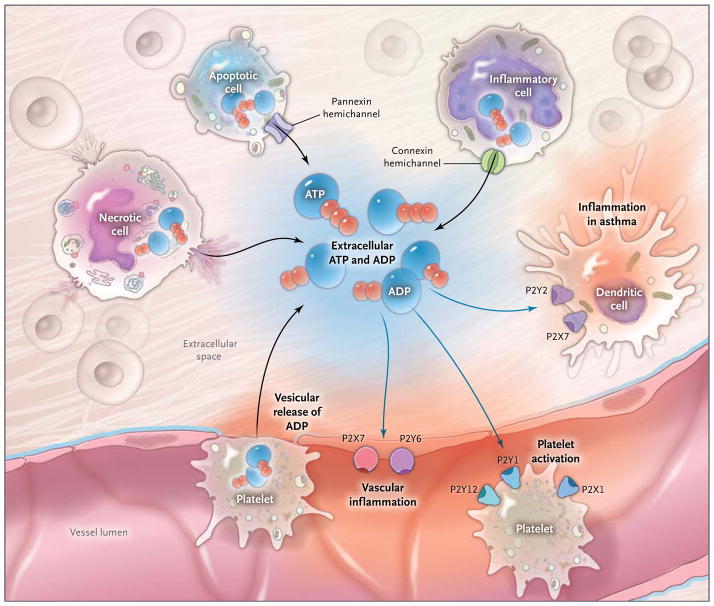
During inflammatory conditions that occur in vascular thrombosis, hypoxia, ischemia, inflammatory bowel disease, and acute lung injury, multiple cell types release nucleotides, typically in the form of ATP or ADP, from the intracellular compartment into the extracellular space. The release of nucleotides includes release of ATP from necrotic cells, pannexin-hemichannel– dependent release of ATP during apoptosis, and release of ATP through connexin hemichannels from activated inflammatory cells such as polymorphonuclear granulocytes (neutrophils).,, In addition, release of extracellular ATP has been shown to occur through vesicular exocytosis or connexin hemichannels from endothelial and urothelial cells, osteoblasts, and astrocytes, as well as nerves (not shown). An additional source of extracellular nucleotides in inflammatory conditions is provided by activated platelets, which release ATP and ADP through the release of granules and exocytosis. In the extracellular space, these nucleotides function as signaling molecules that can activate P2Y receptors (G-protein– coupled receptors) or P2X receptors (ligand-gated ion channels). Examples of nucleotide-receptor signaling in inflammatory conditions include P2Y6- or P2X7-receptor signaling, which mediates vascular inflammation,, and P2Y1-, P2X1-, and P2Y12-receptor signaling, which mediates platelet activation. Activation of P2 receptors of the P2Y2 and P2X7 family that are expressed on dendritic cells is thought to play a role in promoting lung inflammation in chronic lung diseases such as asthma.
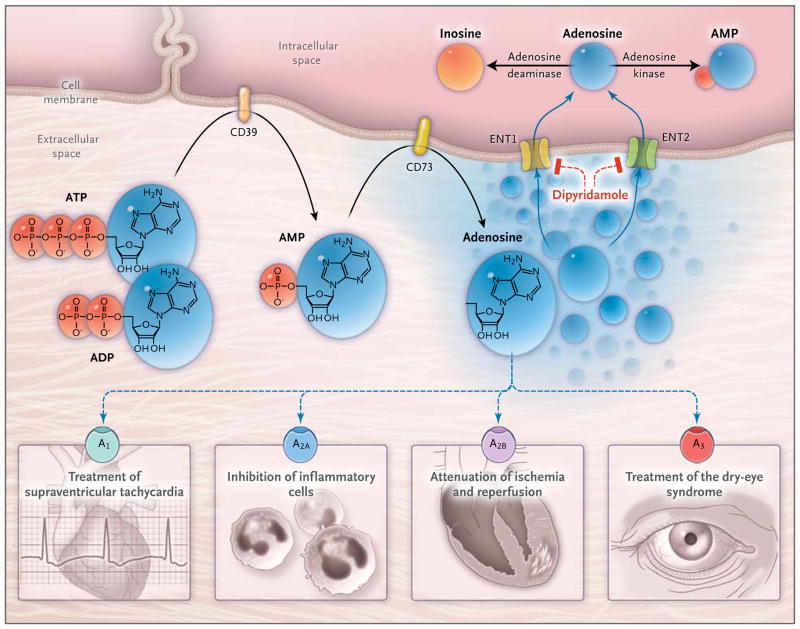
In inflammatory conditions, extracellular adenosine is derived predominantly from the enzymatic conversion of the precursor nucleotides ATP and ADP to AMP through the enzymatic activity of the ectonucleoside triphosphate diphosphohydrolase 1 (CD39) and the subsequent conversion of AMP to adenosine through ecto-5′-nucleotidase (CD73). Extracellular adenosine can signal through four distinct adenosine receptors: ADORA1 (A1), ADORA2A (A2A), ADORA2B (A2B), and ADORA3 (A3). An example of the functional role of extracellular adenosine signaling is A1-receptor activation during intravenous administration of adenosine for the treatment of supraventricular tachycardia. In addition, experimental studies implicate activation of A2A that is expressed on inflammatory cells such as neutrophils or lymphocytes in the attenuation of inflammation., Other experimental studies provide evidence of signaling events through A2B in tissue adaptation to hypoxia and attenuation of ischemia and reperfusion.– A clinical trial has shown that an oral agonist of the A3 adenosine receptor may be useful in the treatment of the dry-eye syndrome. Adenosine signaling is terminated by adenosine uptake from the extracellular space toward the intracellular space, predominantly through equilibrative nucleoside transporter 1 (ENT1) and equilibrative nucleoside transporter 2 (ENT2), followed by metabolism of adenosine to AMP through the adenosine kinase or to inosine through the adenosine deaminase. Blockade of equilibrative nucleoside transporters by dipyridamole is associated with increased extracellular adenosine concentrations and signaling (e.g., during pharmacologic stress echocardiography or in protection of tissue from ischemia).
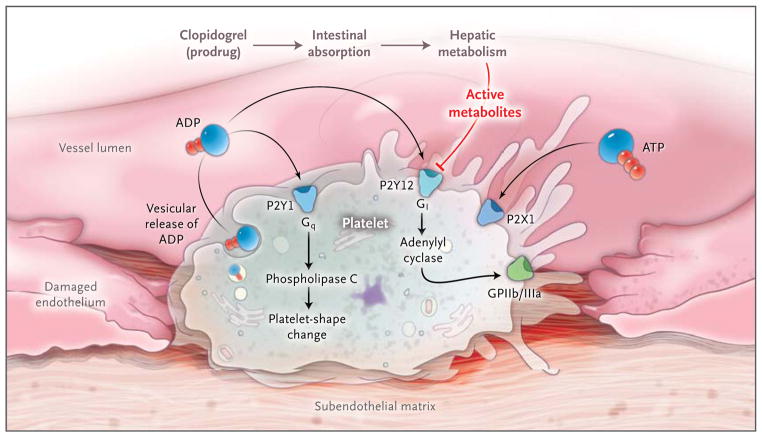
Purinergic signaling is an important link among platelet activation, vascular thrombosis, and inflammation. Platelets mediate primary hemostasis through adhesion, aggregation, and subsequent thrombus formation. Extracellular nucleotides are continuously released from cells associated with the blood. When the vessel wall is injured, platelets roll and become tethered to the subendothelial matrix. These interactions cause changes in the shape of platelets and vesicular release of ADP. Platelet responses to ADP require the coordinated activation of P2Y1 and P2Y12, which function as guanosine triphosphate–binding protein (G protein)-coupled receptors. ADP-dependent activation of the P2Y1 receptor causes activation of phospholipase C mediated through the G protein Gq, leading to subsequent changes in the shape of platelets. ADP also activates P2Y12 receptors, with subsequent activation of the G protein Gi, thereby contributing to fibrinogen-receptor activation (the glycoprotein IIb/IIIa [GPIIb/IIIa] receptor) and platelet aggregation. The platelet inhibitor clopidogrel has pharmacologic effects on the P2Y12 receptor once it has been absorbed and converted from a prodrug to active metabolites. Both intestinal absorption of the prodrug and hepatic metabolism are controlled by specific gene products. Active clopidogrel metabolites irreversibly antagonize the P2Y12 receptor, which in turn inactivates the GPIIb/IIIa-fibrinogen receptor. Clinical trials that examined genetic determinants of the response to clopidogrel and cardiovascular events indicate that gene mutations affecting drug absorption and metabolic activation, rather than P2Y12 mutations, account for the pharmacologic effects of clopidogrel. Platelets also express P2X1 receptors, which mediate modulation of platelet aggregation and shape changes after activation by ADP or ATP in vivo.
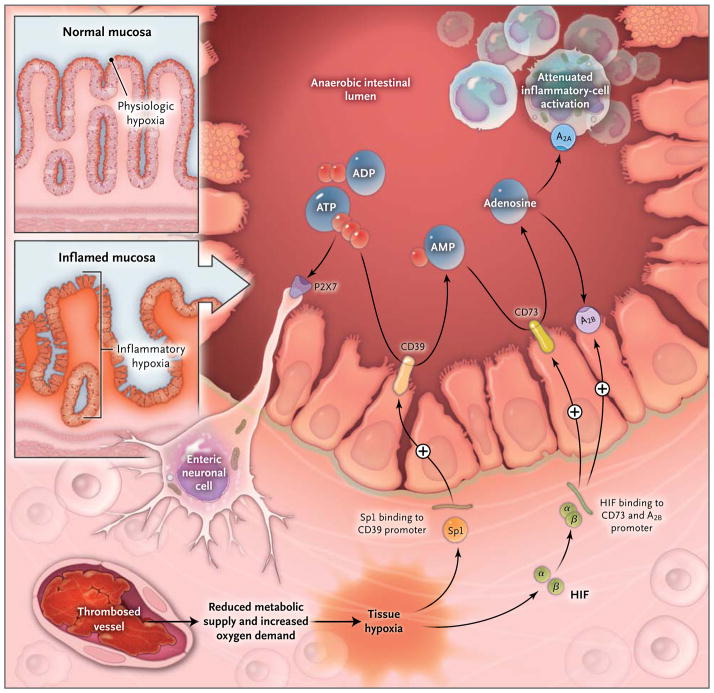
Histologic staining of intestinal sections for hypoxia shows that hypoxia is present within the apical surface of the intestinal mucosa (orange area in upper insert). This presence is most likely due to the fact that the intestinal lumen is anaerobic, which results in a steep oxygen gradient across the epithelial monolayer. In patients with intestinal inflammation such as that which occurs in the course of inflammatory bowel disease, a decrease in metabolic supply (e.g., due to thrombosed vessels) and profound increases in oxygen demand result in an imbalance in oxygen availability. This imbalance causes severe hypoxia of the inflamed mucosa, as indicated by histologic staining for tissue hypoxia (as shown in the lower insert, the orange staining that extends from the apical aspects of the mucosa into the crypts and submucosal tissues indicates severe tissue hypoxia). Release of ATP or ADP from inflammatory cells, platelets, or epithelial cells results in the activation of P2 receptors such as the P2X7 receptor, expressed on enteric neurons, thereby promoting tissue inflammation and injury. Hypoxia causes the activation of transcriptional programs that result in an Sp1-dependent induction of CD39 and a hypoxia-inducible factor (HIF)–dependent induction of CD73 and the ADORA2B (A2B) adenosine receptor. These transcriptional changes lead to an increased rate of turnover of the extracellular nucleotides ATP and ADP to AMP (through CD39) and subsequently to adenosine (through CD73). Experimental studies indicate that adenosine-receptor activation — particularly through Adora2a (A2A) and A2B
— dampens intestinal inflammation and promotes epithelial integrity during intestinal inflammation.
Comment in
-
Purinergic signaling during inflammation.N Engl J Med. 2013 Mar 28;368(13):1260. doi: 10.1056/NEJMc1300259. N Engl J Med. 2013. PMID: 23534573 No abstract available.
-
Purinergic signaling during inflammation.N Engl J Med. 2013 Mar 28;368(13):1260. doi: 10.1056/NEJMc1300259. N Engl J Med. 2013. PMID: 23534574 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources