

Automated design of ligands to polypharmacological profiles
- PMID: 23235874
- PMCID: PMC3653568
- DOI: 10.1038/nature11691
Automated design of ligands to polypharmacological profiles
Abstract
The clinical efficacy and safety of a drug is determined by its activity profile across many proteins in the proteome. However, designing drugs with a specific multi-target profile is both complex and difficult. Therefore methods to design drugs rationally a priori against profiles of several proteins would have immense value in drug discovery. Here we describe a new approach for the automated design of ligands against profiles of multiple drug targets. The method is demonstrated by the evolution of an approved acetylcholinesterase inhibitor drug into brain-penetrable ligands with either specific polypharmacology or exquisite selectivity profiles for G-protein-coupled receptors. Overall, 800 ligand-target predictions of prospectively designed ligands were tested experimentally, of which 75% were confirmed to be correct. We also demonstrate target engagement in vivo. The approach can be a useful source of drug leads when multi-target profiles are required to achieve either selectivity over other drug targets or a desired polypharmacology.
Figures
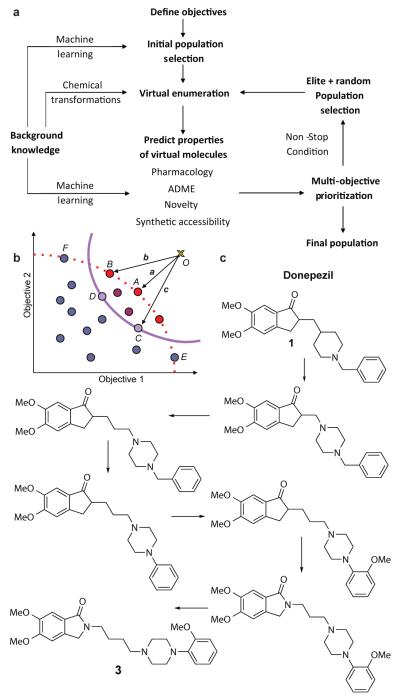
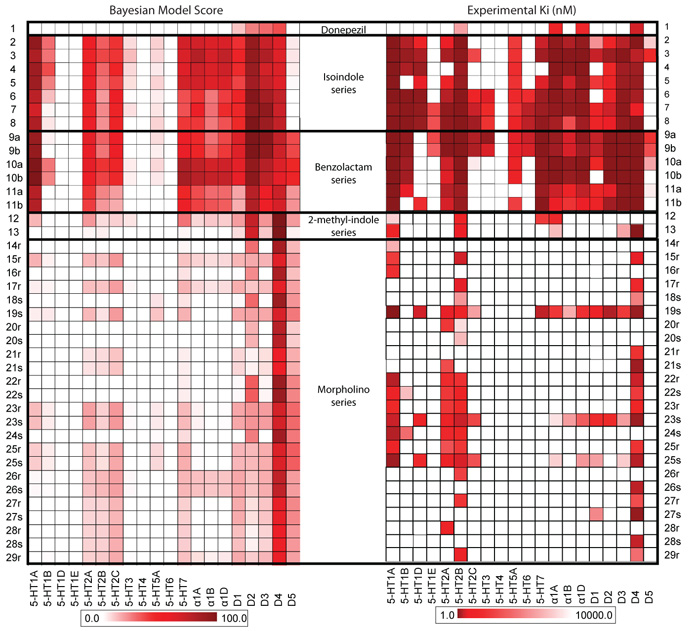
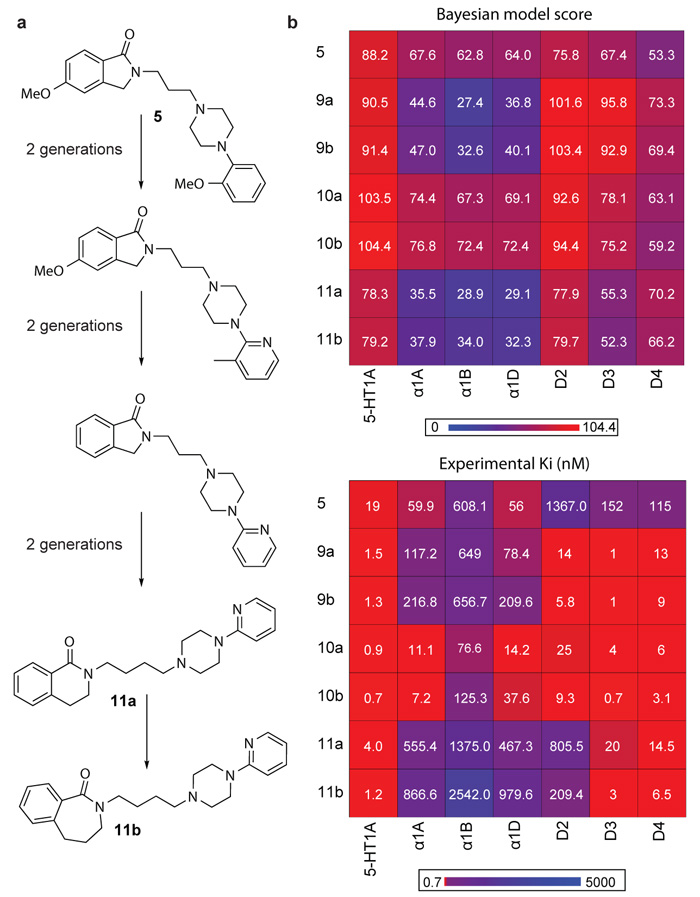
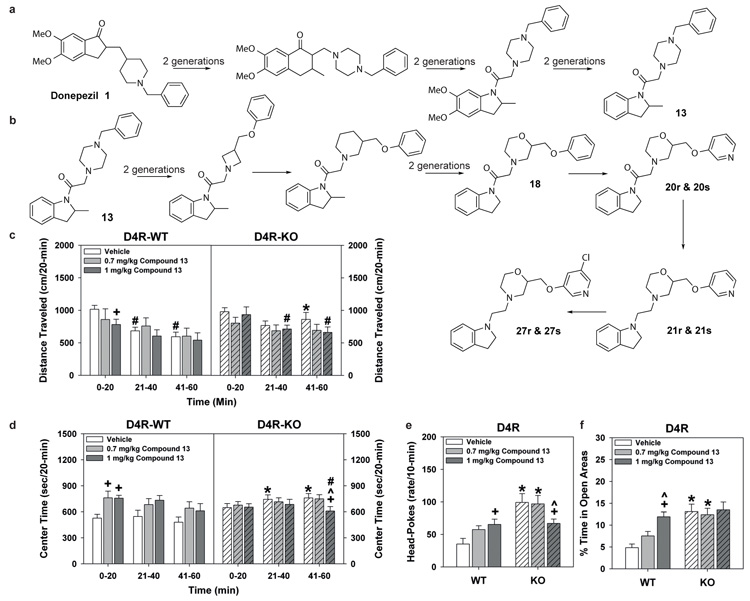
Comment in
-
Computational chemistry: Homing in on desired drug properties.Nat Rev Drug Discov. 2013 Feb;12(2):101. doi: 10.1038/nrd3938. Nat Rev Drug Discov. 2013. PMID: 23370243 No abstract available.
References
-
- Hughes JD, et al. Physicochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008;18:4872–4875. - PubMed
-
- Roth BL. Drugs and valvular heart disease. N. Engl. J. Med. 2007;356:6–9. - PubMed
-
- Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Drug target identification using side-effect similarity. Science. 2008;321:263–266. - PubMed
-
- Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nature Rev. Drug Discov. 2004;3:353–359. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
