

Neuronal gap junctions: making and breaking connections during development and injury
- PMID: 23237660
- PMCID: PMC3609876
- DOI: 10.1016/j.tins.2012.11.001
Neuronal gap junctions: making and breaking connections during development and injury
Abstract
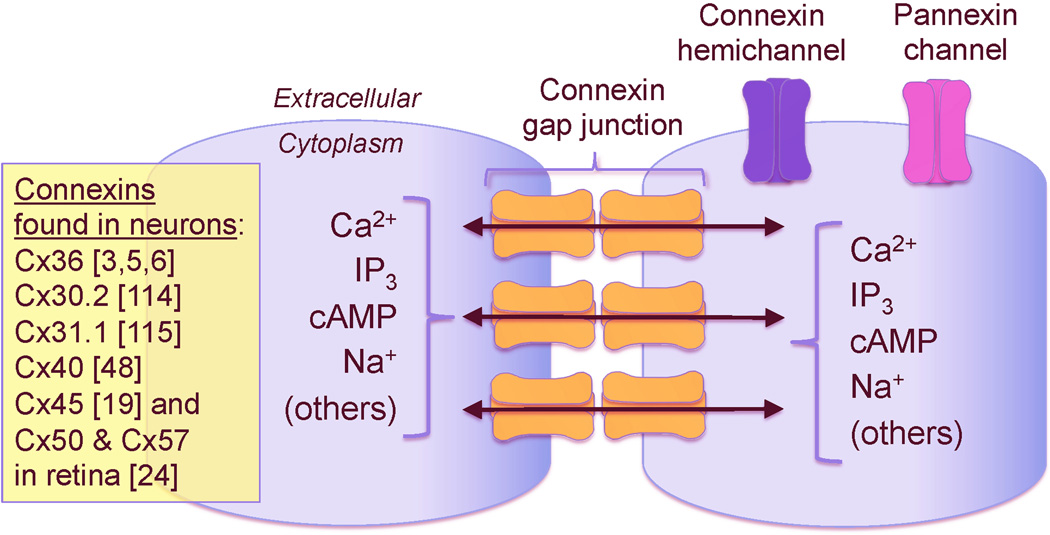
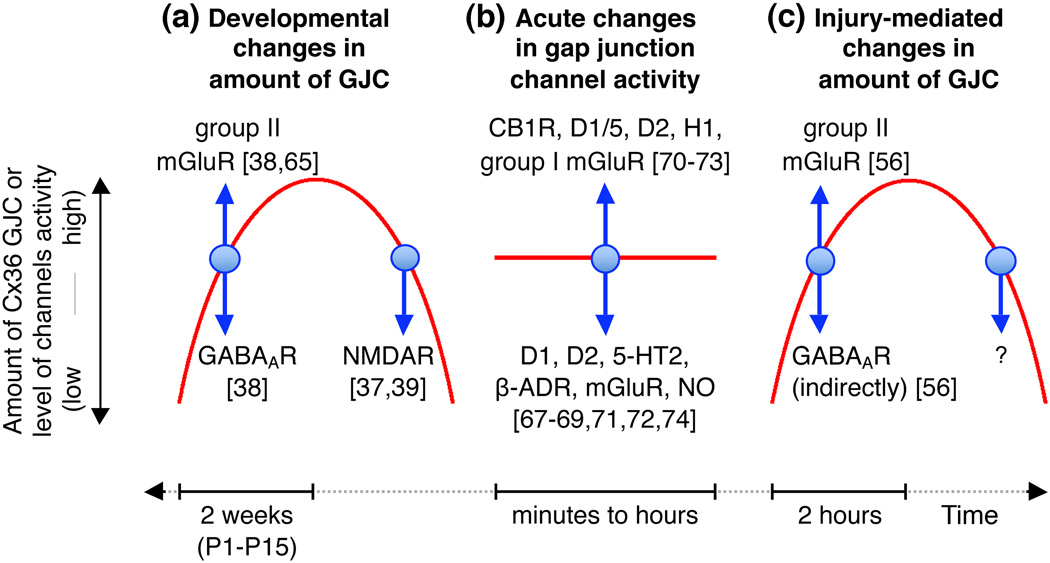
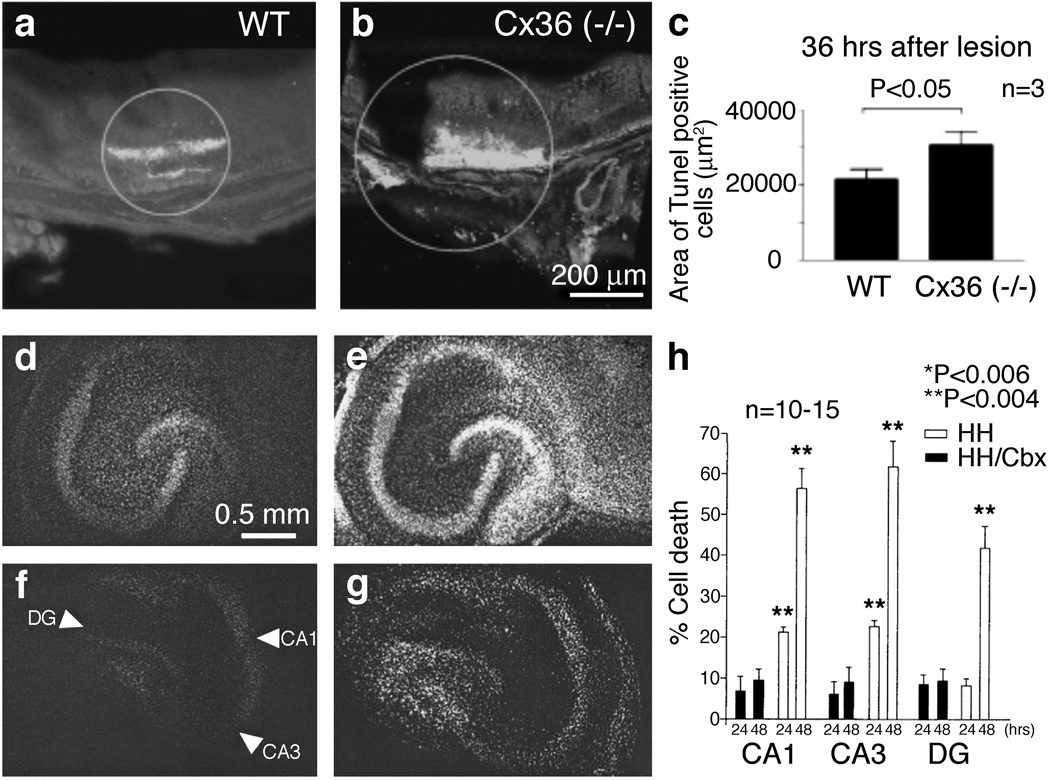
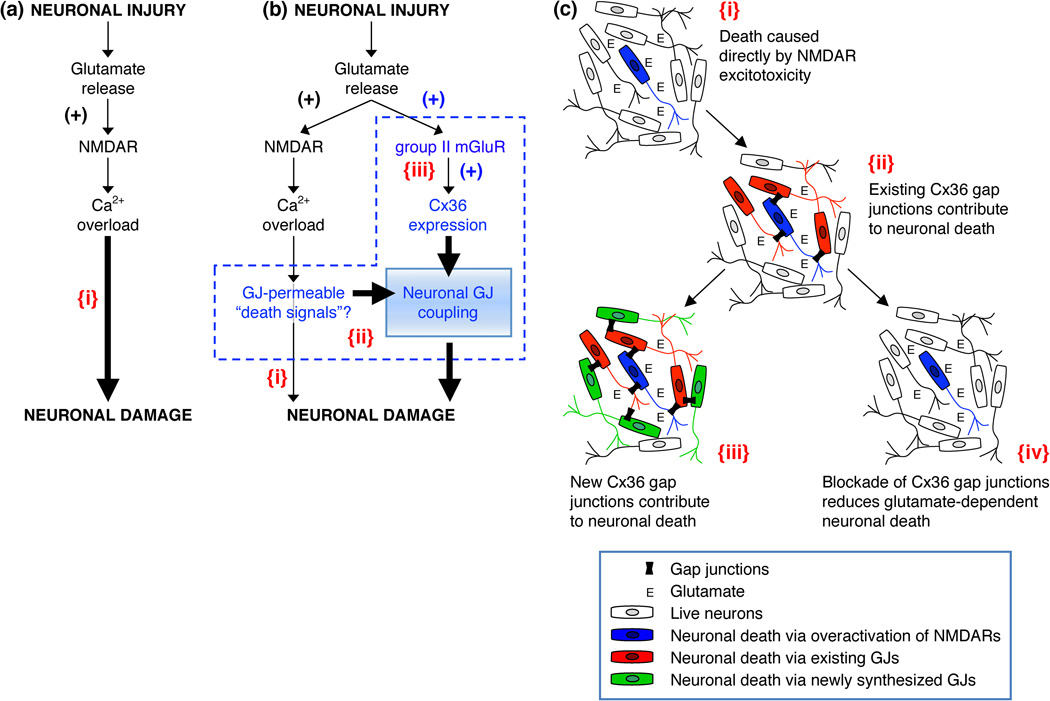
In the mammalian central nervous system (CNS), coupling of neurons by gap junctions (i.e., electrical synapses) and the expression of the neuronal gap junction protein, connexin 36 (Cx36), transiently increase during early postnatal development. The levels of both subsequently decline and remain low in the adult, confined to specific subsets of neurons. However, following neuronal injury [such as ischemia, traumatic brain injury (TBI), and epilepsy], the coupling and expression of Cx36 rise. Here we summarize new findings on the mechanisms of regulation of Cx36-containing gap junctions in the developing and mature CNS and following injury. We also review recent studies suggesting various roles for neuronal gap junctions and in particular their role in glutamate-mediated neuronal death.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
References
-
- Beyer EC, Berthoud VM. Chapter 1: The family of connexin genes. In: Harris A, Locke D, editors. Connexins: A Guide. Humana Press; 2009. pp. 3–26.
-
- Giaume C, Theis M. Pharmacological and genetic approaches to study connexin-mediated channels in glial cells of the central nervous system. Brain Res Rev. 2010;63:160–176. - PubMed
-
- Belluardo N, et al. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865:121–138. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
