

A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis
- PMID: 23243209
- PMCID: PMC3574639
- DOI: 10.1161/CIRCULATIONAHA.112.116988
A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis
Abstract
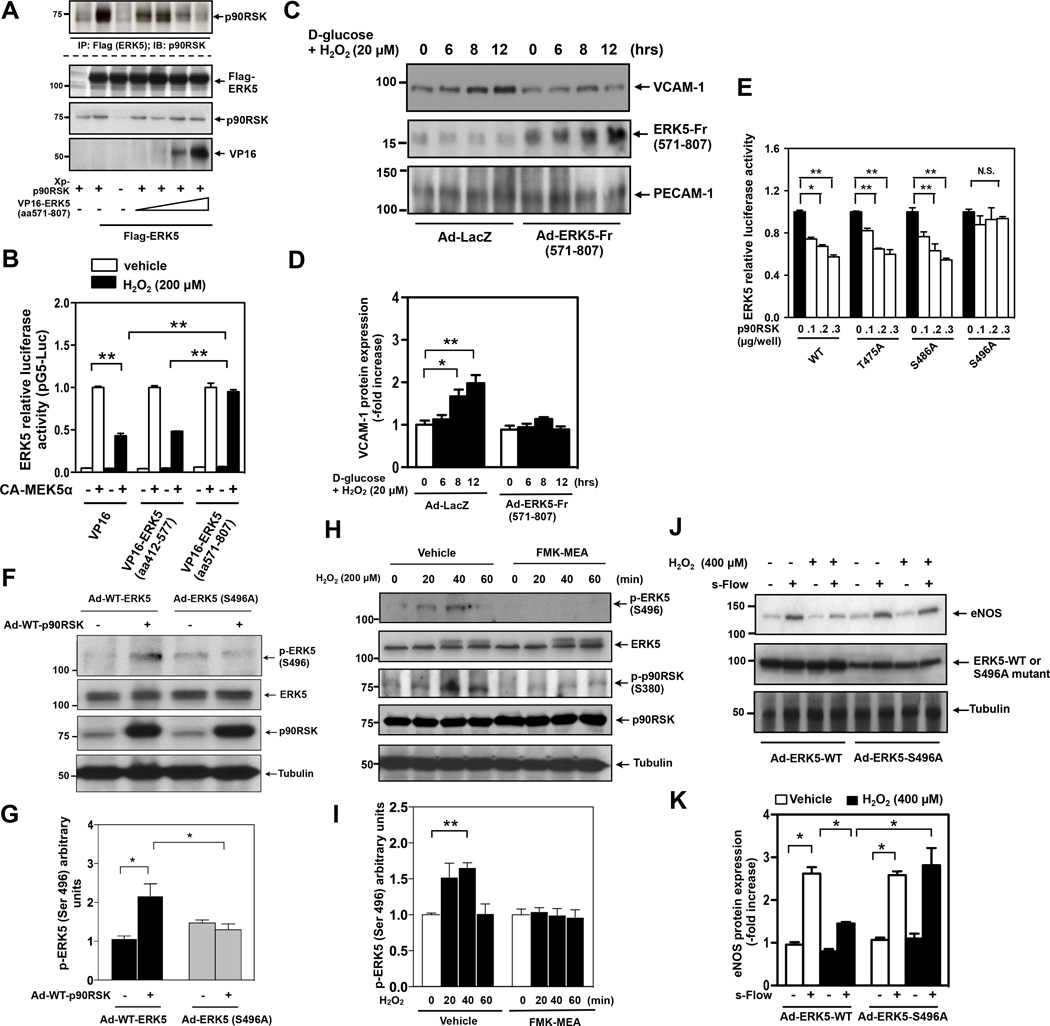
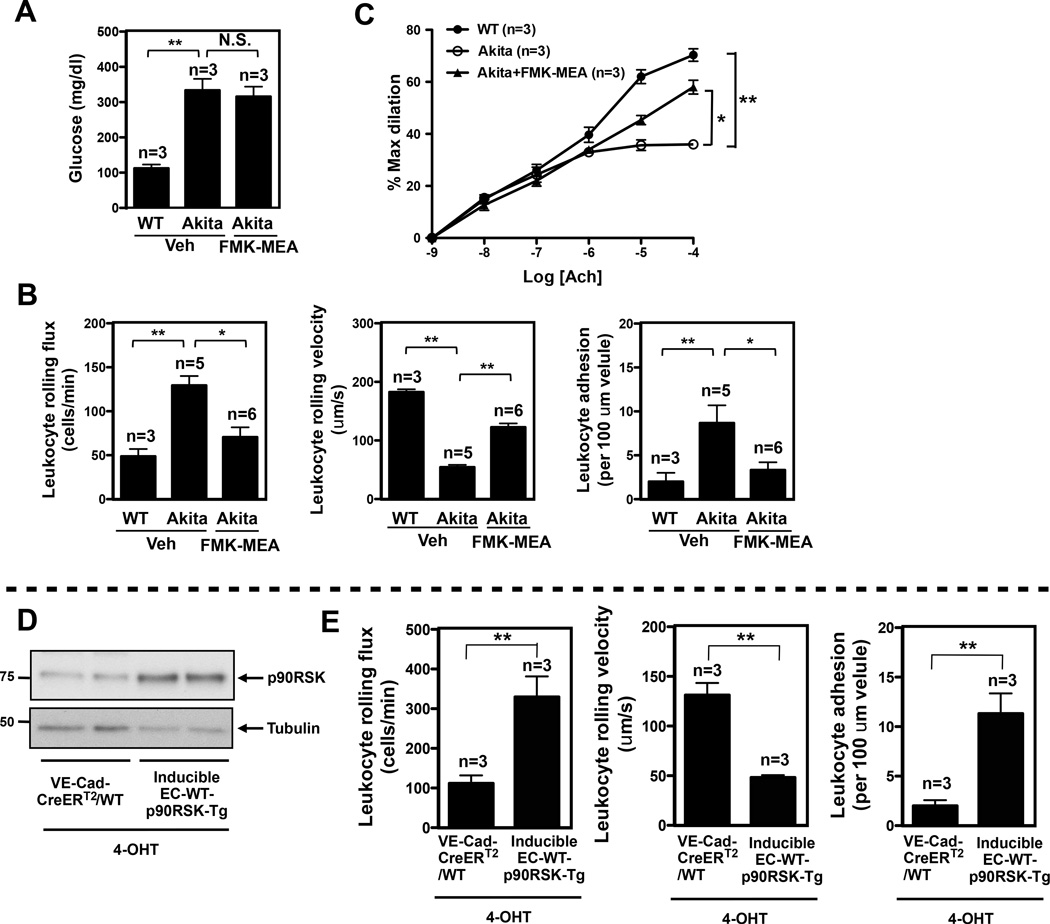
Background: Diabetes mellitus is a major risk factor for cardiovascular mortality by increasing endothelial cell (EC) dysfunction and subsequently accelerating atherosclerosis. Extracellular-signal regulated kinase 5 (ERK5) is activated by steady laminar flow and regulates EC function by increasing endothelial nitric oxide synthase expression and inhibiting EC inflammation. However, the role and regulatory mechanisms of ERK5 in EC dysfunction and atherosclerosis are poorly understood. Here, we report the critical role of the p90 ribosomal S6 kinase (p90RSK)/ERK5 complex in EC dysfunction in diabetes mellitus and atherosclerosis.
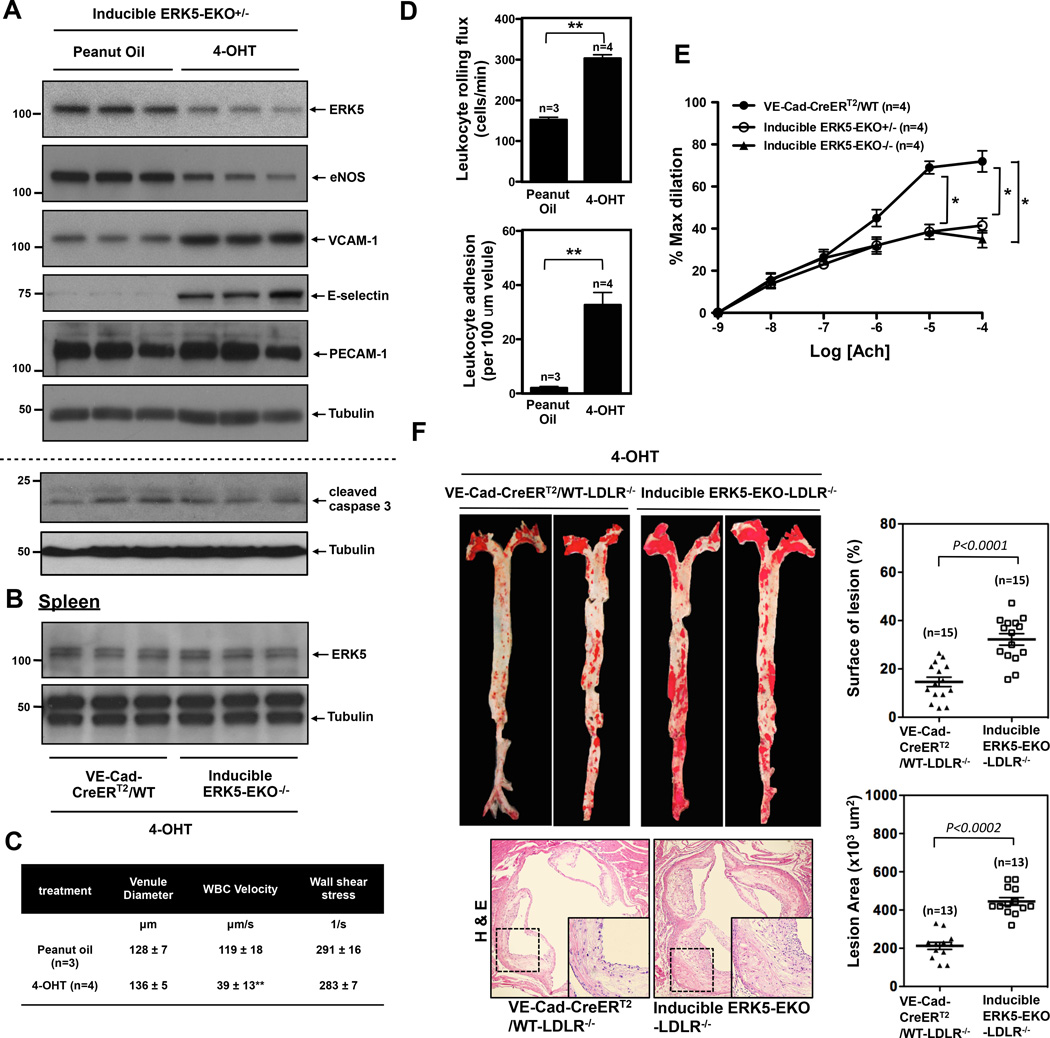
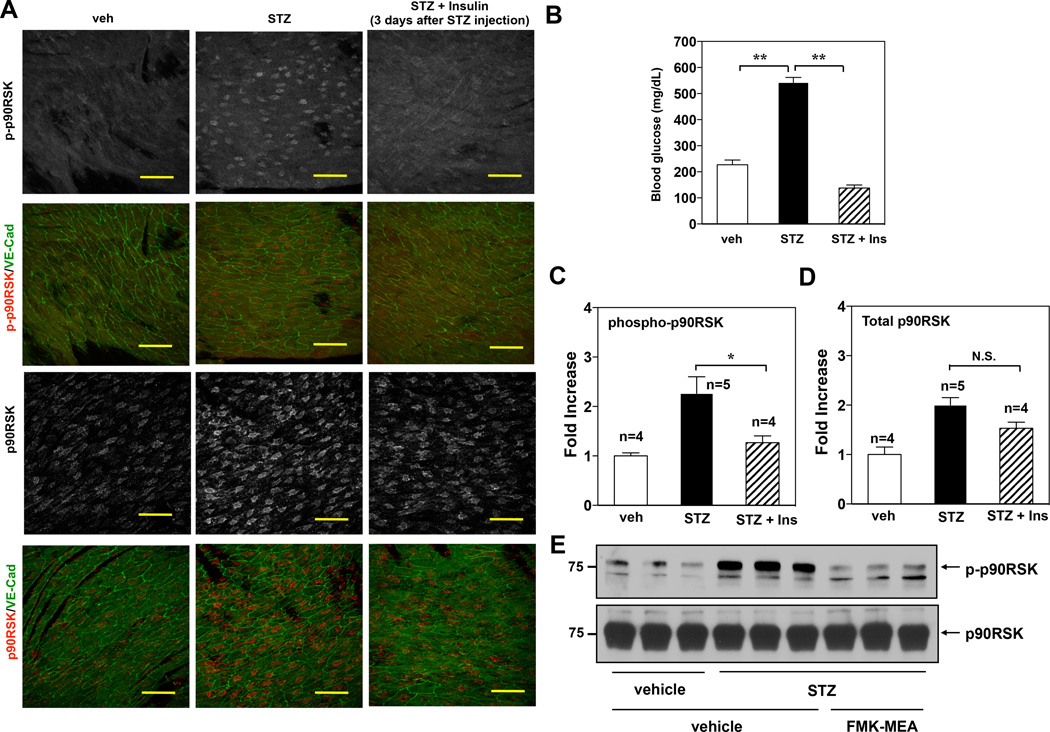
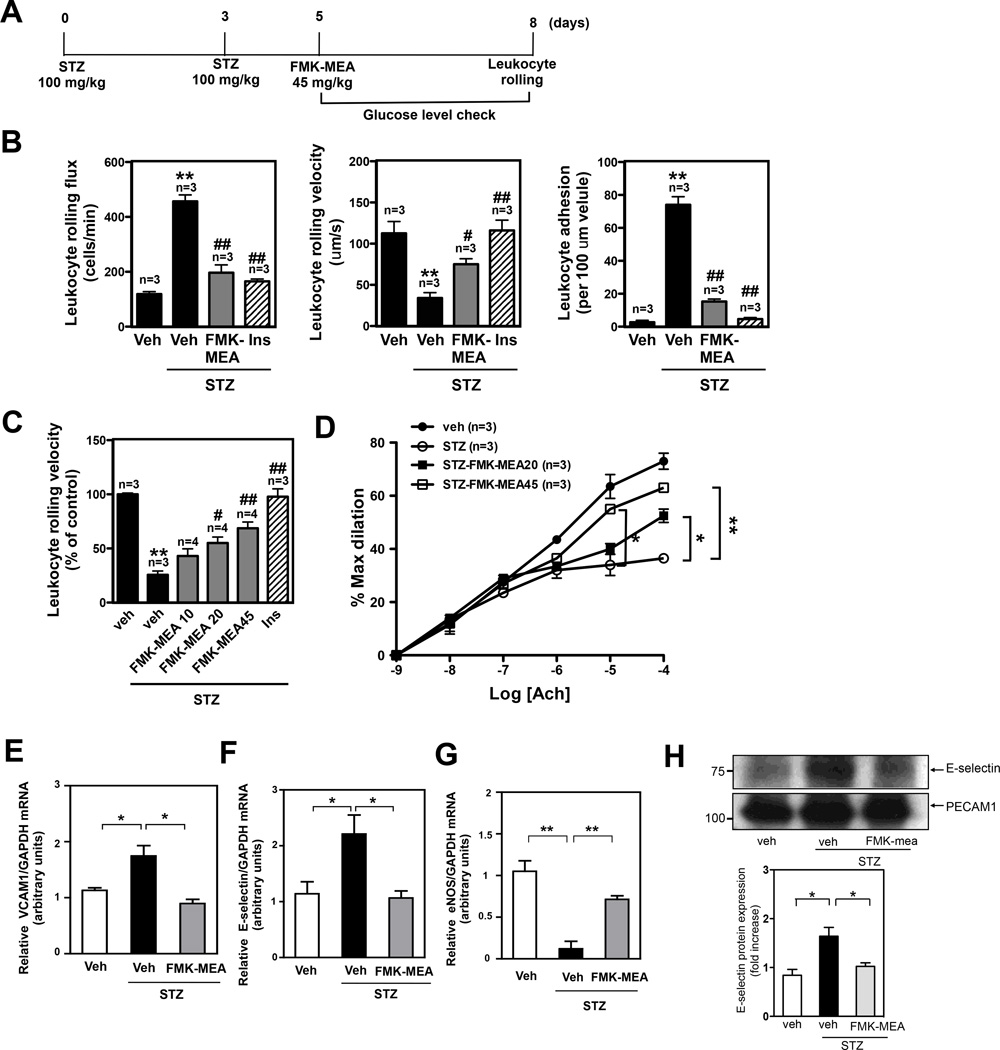
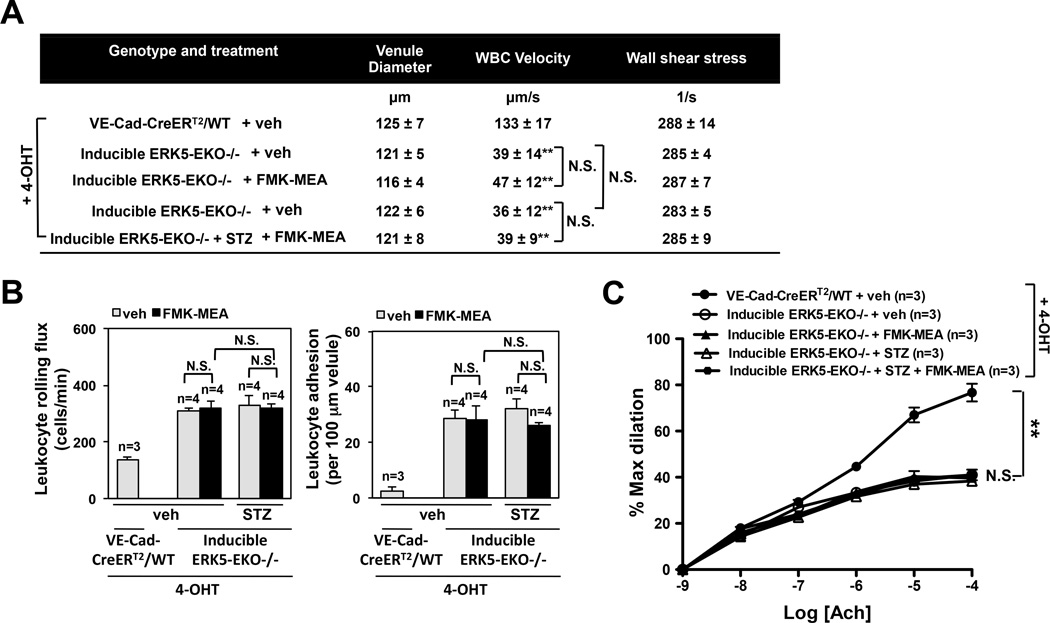
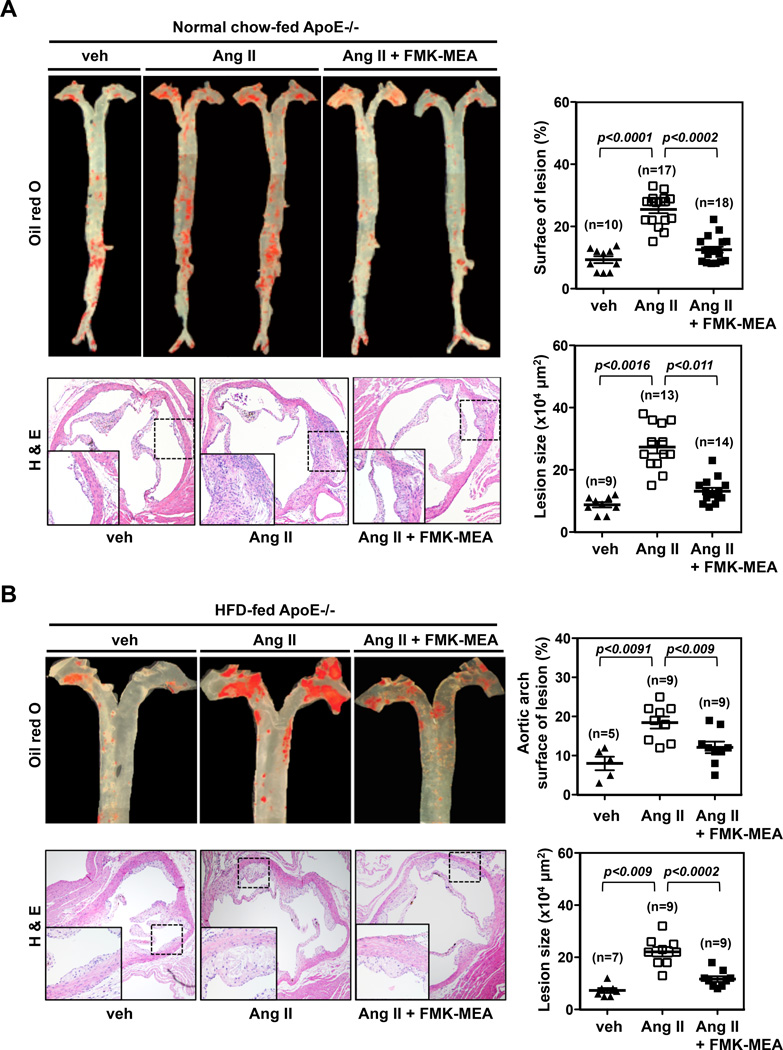
Methods and results: Inducible EC-specific ERK5 knockout (ERK5-EKO) mice showed increased leukocyte rolling and impaired vessel reactivity. To examine the role of endothelial ERK5 in atherosclerosis, we used inducible ERK5-EKO-LDLR(-/-) mice and observed increased plaque formation. When activated, p90RSK associated with ERK5, and this association inhibited ERK5 transcriptional activity and upregulated vascular cell adhesion molecule 1 expression. In addition, p90RSK directly phosphorylated ERK5 S496 and reduced endothelial nitric oxide synthase expression. p90RSK activity was increased in diabetic mouse vessels, and fluoromethyl ketone-methoxyethylamine, a specific p90RSK inhibitor, ameliorated EC-leukocyte recruitment and diminished vascular reactivity in diabetic mice. Interestingly, in ERK5-EKO mice, increased leukocyte rolling and impaired vessel reactivity were resistant to the beneficial effects of fluoromethyl ketone-methoxyethylamine, suggesting a critical role for endothelial ERK5 in mediating the salutary effects of fluoromethyl ketone-methoxyethylamine on endothelial dysfunction. Fluoromethyl ketone-methoxyethylamine also inhibited atherosclerosis formation in ApoE(-/-) mice.
Conclusions: Our study highlights the importance of the p90RSK/ERK5 module as a critical mediator of EC dysfunction in diabetes mellitus and atherosclerosis formation, thus revealing a potential new target for therapeutic intervention.
Conflict of interest statement
Figures

References
-
- Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. - PubMed
-
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ Res. 2000;87:840–844. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL-088637/HL/NHLBI NIH HHS/United States
- R01 HL111480/HL/NHLBI NIH HHS/United States
- R01 GM071434/GM/NIGMS NIH HHS/United States
- R01 HL108551/HL/NHLBI NIH HHS/United States
- HL-077789/HL/NHLBI NIH HHS/United States
- R01 HL064839/HL/NHLBI NIH HHS/United States
- PG/12/76/29852/BHF_/British Heart Foundation/United Kingdom
- GM-071434/GM/NIGMS NIH HHS/United States
- R01 HL102746/HL/NHLBI NIH HHS/United States
- UL1 RR024160/RR/NCRR NIH HHS/United States
- PG/09/052/27833/BHF_/British Heart Foundation/United Kingdom
- P01 HL077789/HL/NHLBI NIH HHS/United States
- HL-064839/HL/NHLBI NIH HHS/United States
- R01 HL088637/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
