

The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis
- PMID: 23245857
- PMCID: PMC3594559
- DOI: 10.1016/j.tig.2012.11.005
The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis
Abstract
Recent surveillance data of multidrug-resistant tuberculosis (MDR-TB) reported the highest rates of resistance ever documented. As further amplification of resistance in MDR strains of Mycobacterium tuberculosis occurs, extensively drug-resistant (XDR) and totally drug-resistant (TDR) TB are beginning to emerge. Although for the most part, the epidemiological factors involved in the spread of MDR-TB are understood, insights into the bacterial drivers of MDR-TB have been gained only recently, largely owing to novel technologies and research in other organisms. Herein, we review recent findings on how bacterial factors, such as persistence, hypermutation, the complex interrelation between drug resistance and fitness, compensatory evolution, and epistasis affect the evolution of multidrug resistance in M. tuberculosis. Improved knowledge of these factors will help better predict the future trajectory of MDR-TB, and contribute to the development of new tools and strategies to combat this growing public health threat.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
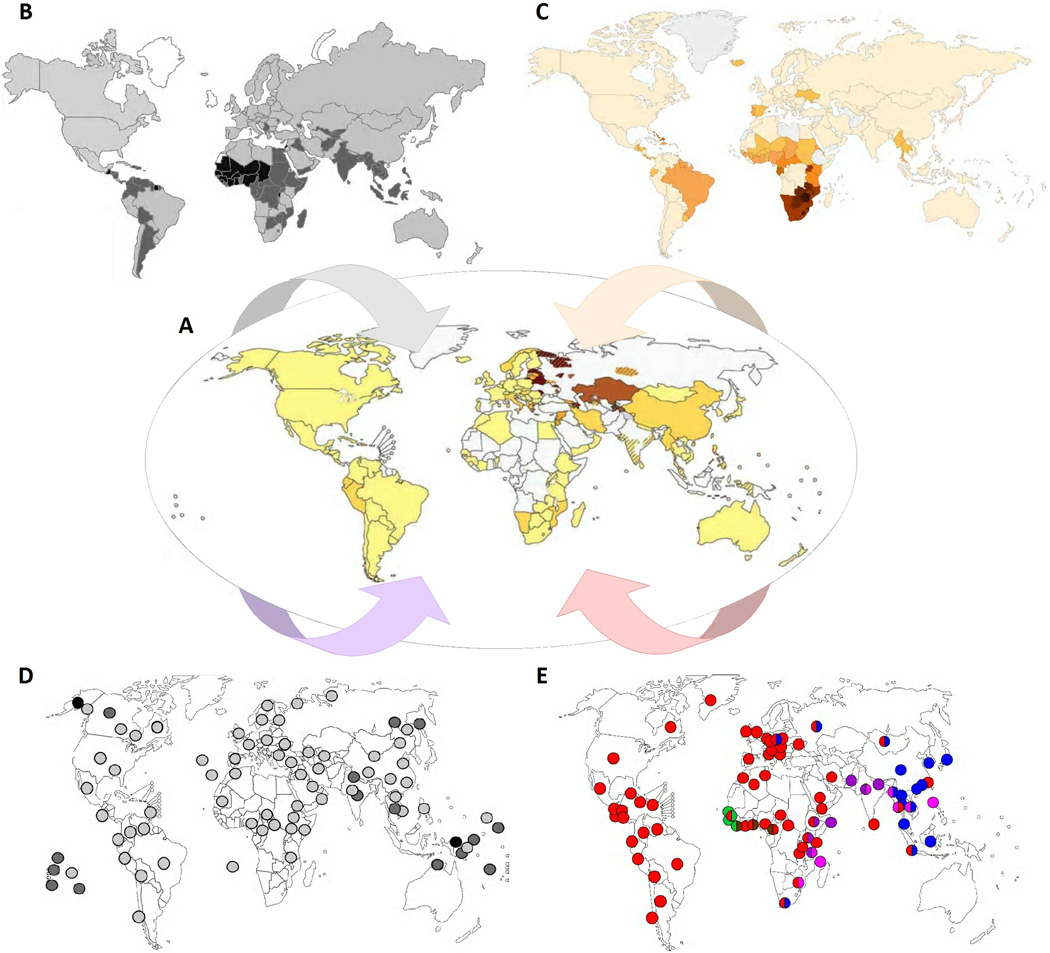
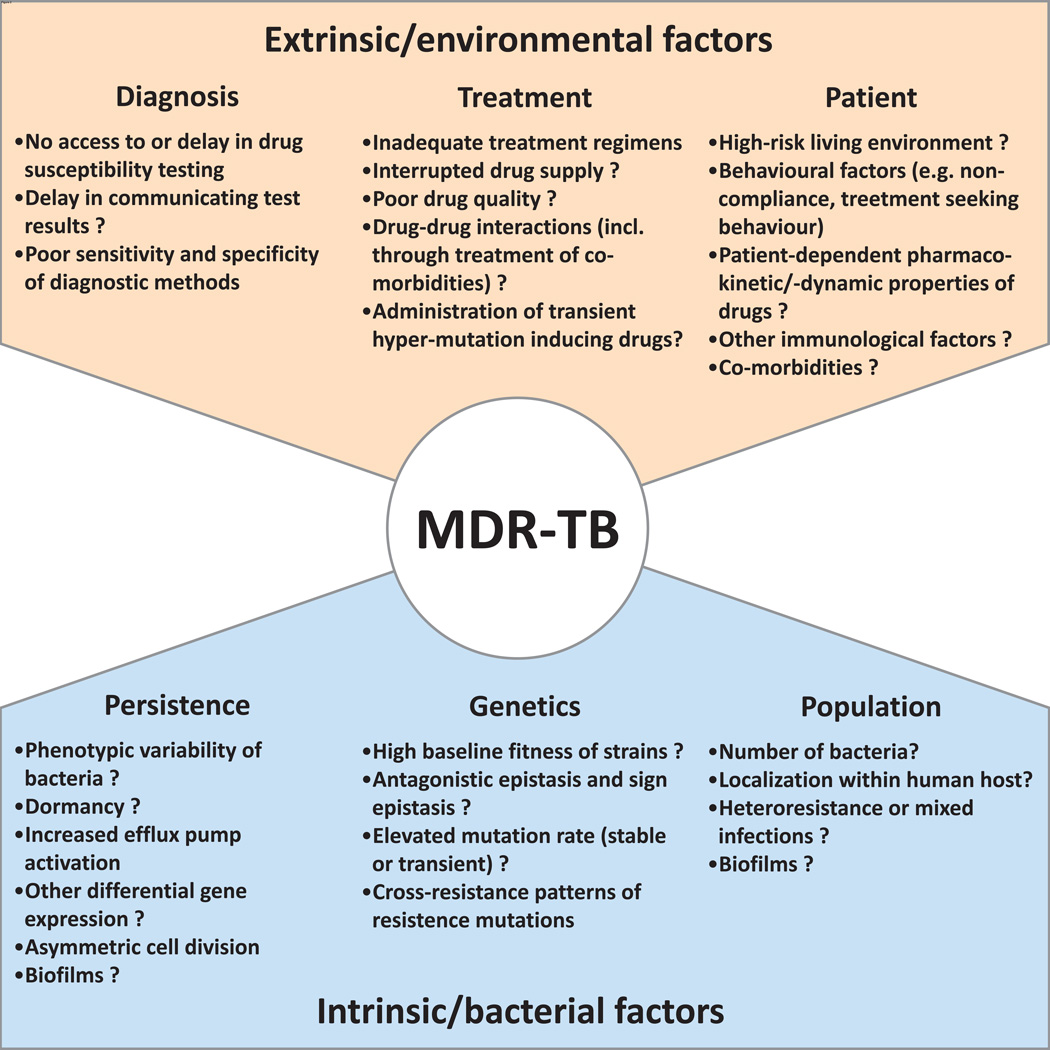
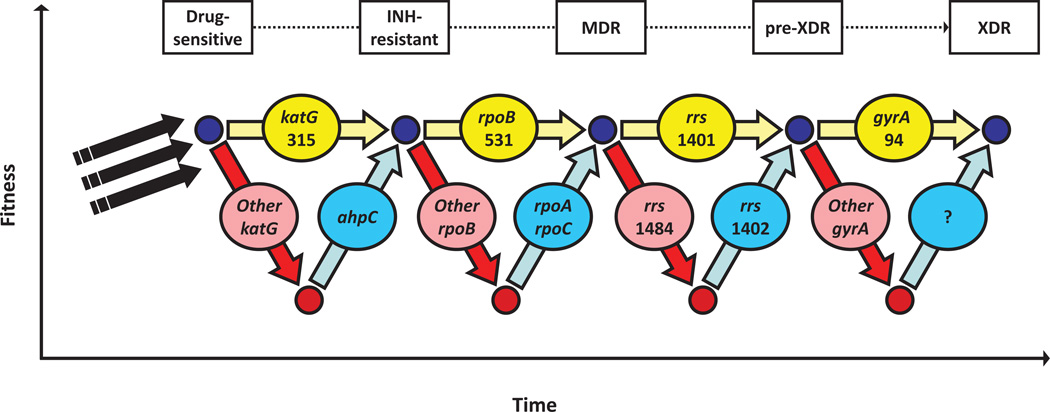
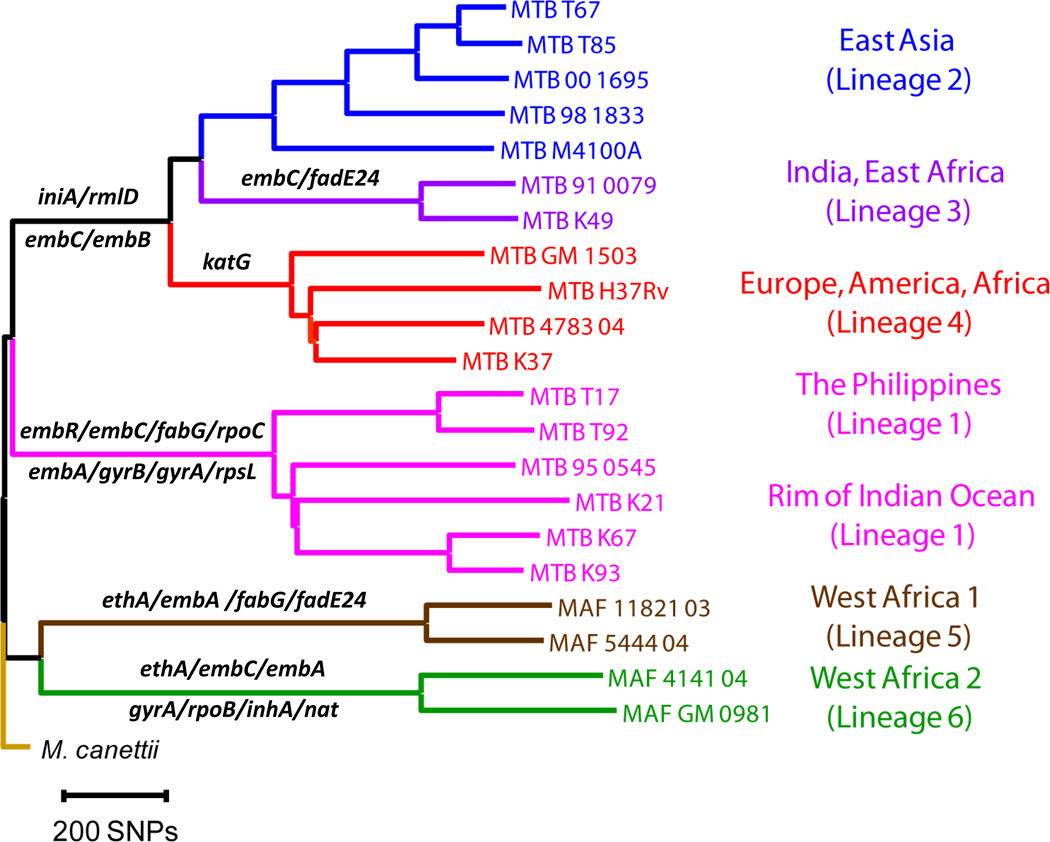
References
-
- World Health Organization. Global tuberculosis control - surveillance, planning, financing. Geneva: World Health Organization; 2012.
-
- Gandhi NR, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet. 2010;375:1830–1843. - PubMed
-
- Caminero JA, et al. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect Dis. 2010;10:621–629. - PubMed
-
- Velayati AA, et al. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest. 2009;136:420–425. - PubMed
-
- Udwadia ZF, et al. Totally drug-resistant tuberculosis in India. Clin Infect Dis. 2012;54:579–581. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
