

Analysis of omics data with genome-scale models of metabolism
- PMID: 23247105
- PMCID: PMC3594511
- DOI: 10.1039/c2mb25453k
Analysis of omics data with genome-scale models of metabolism
Abstract
Over the past decade a massive amount of research has been dedicated to generating omics data to gain insight into a variety of biological phenomena, including cancer, obesity, biofuel production, and infection. Although most of these omics data are available publicly, there is a growing concern that much of these data sit in databases without being used or fully analyzed. Statistical inference methods have been widely applied to gain insight into which genes may influence the activities of others in a given omics data set, however, they do not provide information on the underlying mechanisms or whether the interactions are direct or distal. Biochemically, genetically, and genomically consistent knowledge bases are increasingly being used to extract deeper biological knowledge and understanding from these data sets than possible by inferential methods. This improvement is largely due to knowledge bases providing a validated biological context for interpreting the data.
Figures
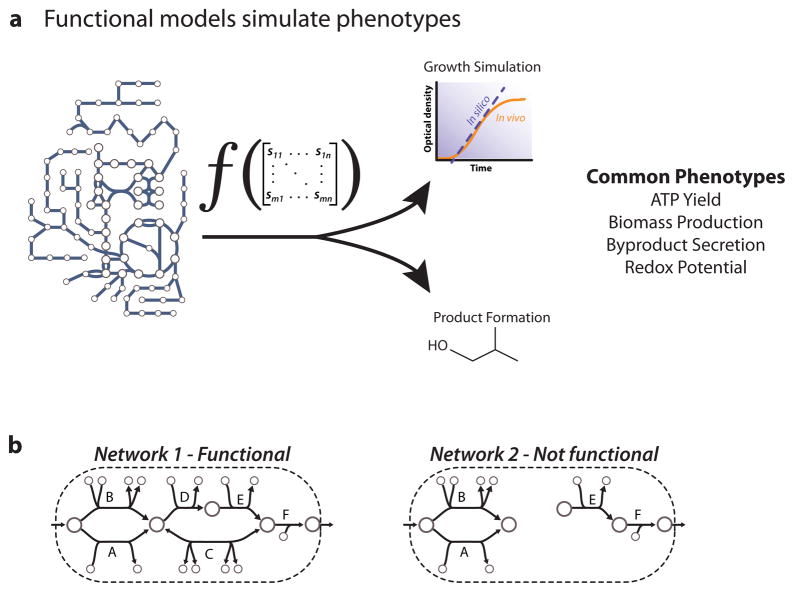
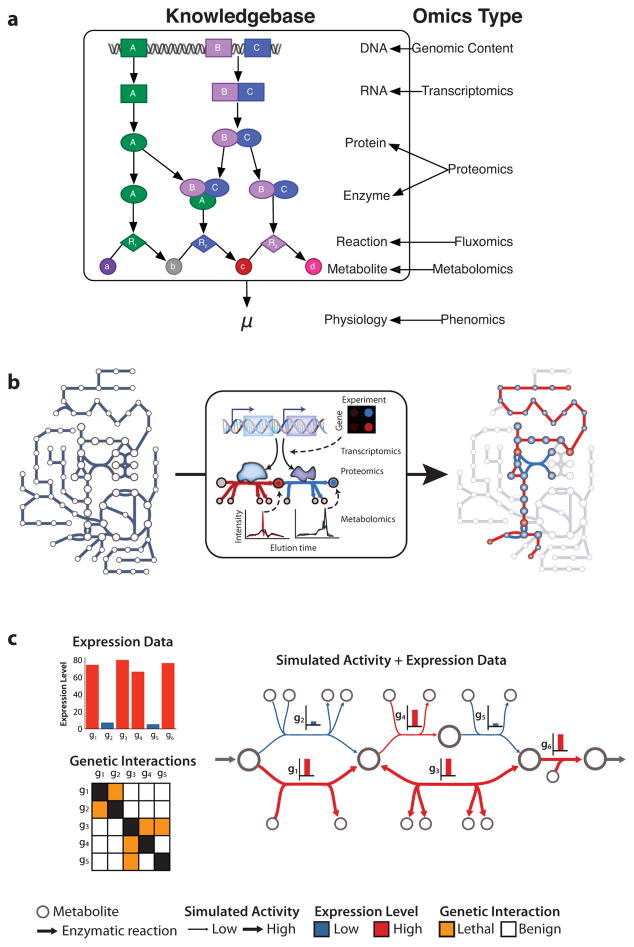
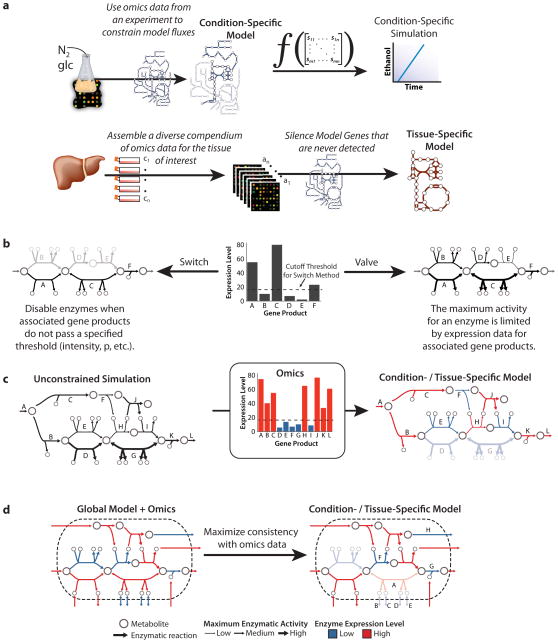
Similar articles
-
Genome-scale model management and comparison.Methods Mol Biol. 2013;985:3-16. doi: 10.1007/978-1-62703-299-5_1. Methods Mol Biol. 2013. PMID: 23417796
-
Enhancement of Plant Productivity in the Post-Genomics Era.Curr Genomics. 2016 Aug;17(4):295-6. doi: 10.2174/138920291704160607182507. Curr Genomics. 2016. PMID: 27499678 Free PMC article.
-
IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures.Front Immunol. 2021 Jul 2;12:687975. doi: 10.3389/fimmu.2021.687975. eCollection 2021. Front Immunol. 2021. PMID: 34276676 Free PMC article.
-
Computational Analysis of Phosphoproteomics Data in Multi-Omics Cancer Studies.Proteomics. 2021 Feb;21(3-4):e1900312. doi: 10.1002/pmic.201900312. Epub 2020 Oct 8. Proteomics. 2021. PMID: 32875713 Review.
-
Integrative analysis of human omics data using biomolecular networks.Mol Biosyst. 2016 Oct 20;12(10):2953-64. doi: 10.1039/c6mb00476h. Epub 2016 Aug 11. Mol Biosyst. 2016. PMID: 27510223 Review.
Cited by
-
Context-Specific Metabolic Model Extraction Based on Regularized Least Squares Optimization.PLoS One. 2015 Jul 9;10(7):e0131875. doi: 10.1371/journal.pone.0131875. eCollection 2015. PLoS One. 2015. PMID: 26158726 Free PMC article.
-
Uncovering the Role of Metabolism in Oomycete-Host Interactions Using Genome-Scale Metabolic Models.Front Microbiol. 2021 Oct 11;12:748178. doi: 10.3389/fmicb.2021.748178. eCollection 2021. Front Microbiol. 2021. PMID: 34707596 Free PMC article. Review.
-
Current advances in systems and integrative biology.Comput Struct Biotechnol J. 2014 Aug 27;11(18):35-46. doi: 10.1016/j.csbj.2014.08.007. eCollection 2014 Aug. Comput Struct Biotechnol J. 2014. PMID: 25379142 Free PMC article. Review.
-
Mapping out the gut microbiota-dependent trimethylamine N-oxide super pathway for systems biology applications.Front Syst Biol. 2023 Mar 8;3:1074749. doi: 10.3389/fsysb.2023.1074749. eCollection 2023. Front Syst Biol. 2023. PMID: 40809486 Free PMC article. Review.
-
Accelerated knowledge discovery from omics data by optimal experimental design.Nat Commun. 2020 Oct 6;11(1):5026. doi: 10.1038/s41467-020-18785-y. Nat Commun. 2020. PMID: 33024104 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous