

Chromatin and the genome integrity network
- PMID: 23247436
- PMCID: PMC3731064
- DOI: 10.1038/nrg3345
Chromatin and the genome integrity network
Abstract
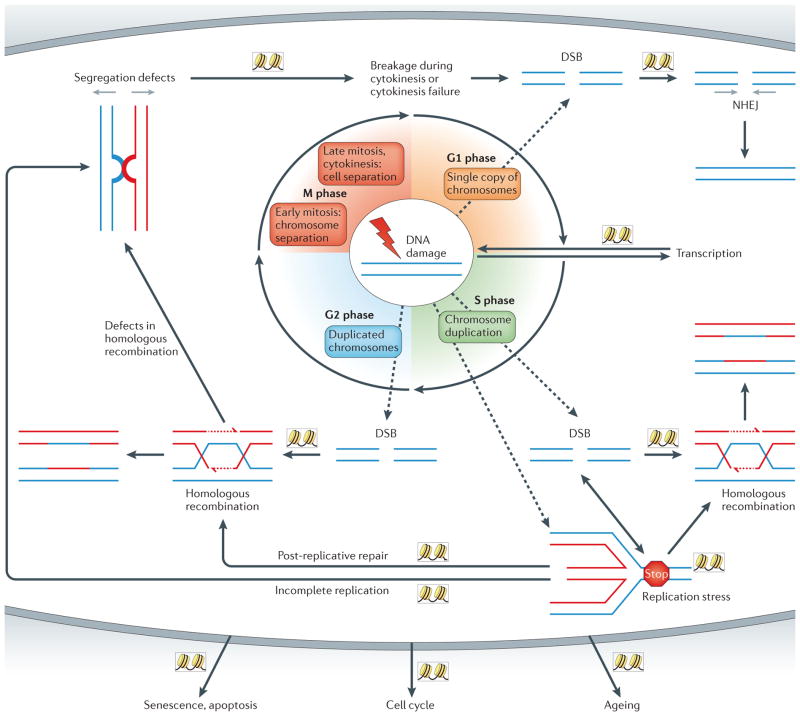
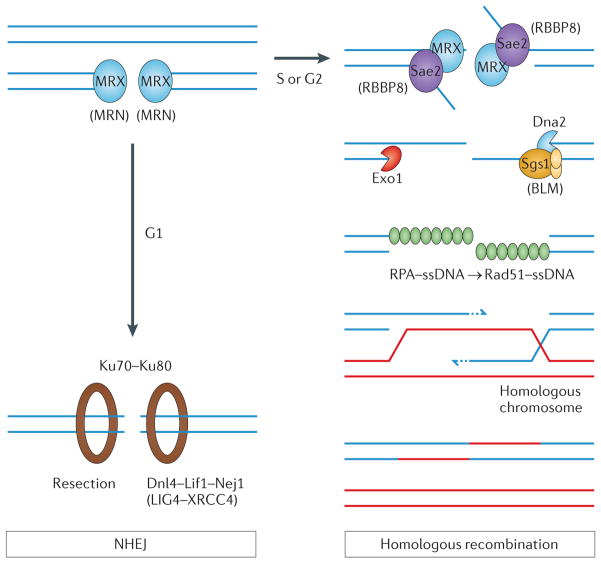
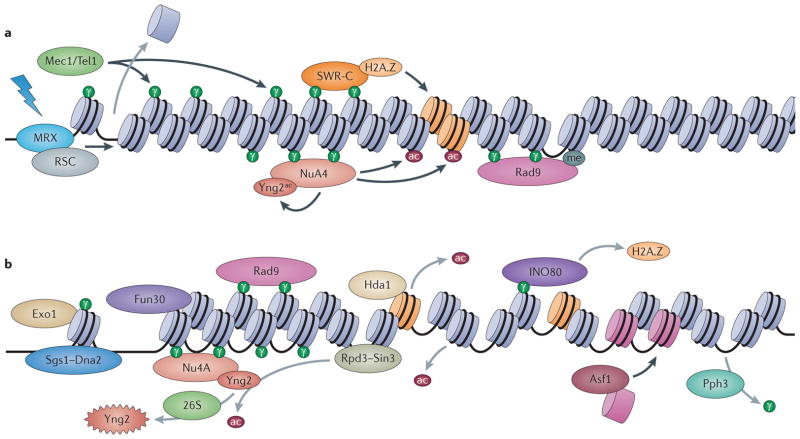
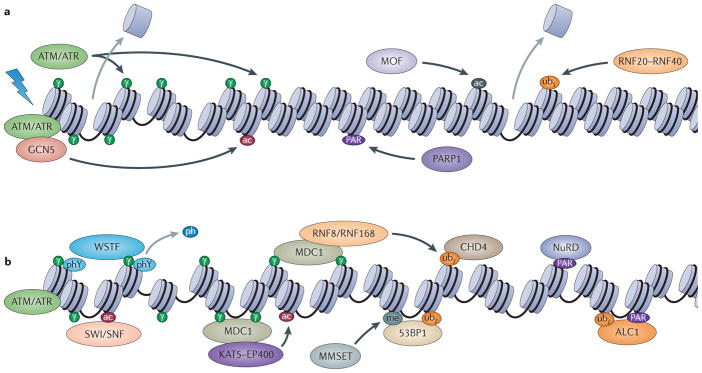
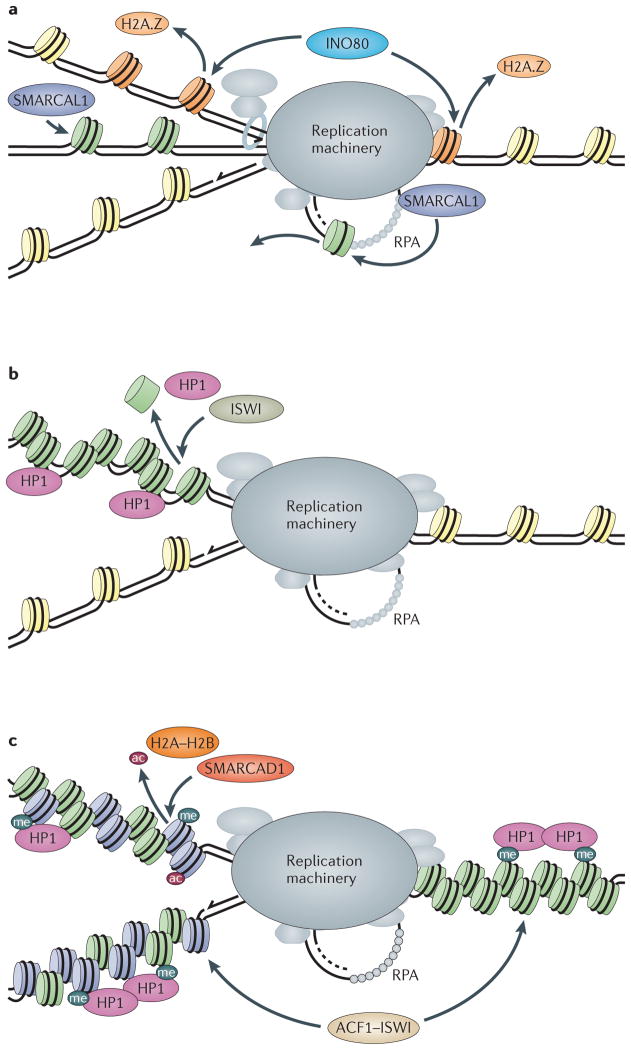
The maintenance of genome integrity is essential for organism survival and for the inheritance of traits to offspring. Genomic instability is caused by DNA damage, aberrant DNA replication or uncoordinated cell division, which can lead to chromosomal aberrations and gene mutations. Recently, chromatin regulators that shape the epigenetic landscape have emerged as potential gatekeepers and signalling coordinators for the maintenance of genome integrity. Here, we review chromatin functions during the two major pathways that control genome integrity: namely, repair of DNA damage and DNA replication. We also discuss recent evidence that suggests a novel role for chromatin-remodelling factors in chromosome segregation and in the prevention of aneuploidy.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nature Rev Mol Cell Biol. 2010;11:208–219. - PubMed
-
- Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nature Rev Genet. 2012;13:189–203. - PubMed
-
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
