

Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2
- PMID: 23248270
- PMCID: PMC3538195
- DOI: 10.1073/pnas.1213680110
Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2
Abstract
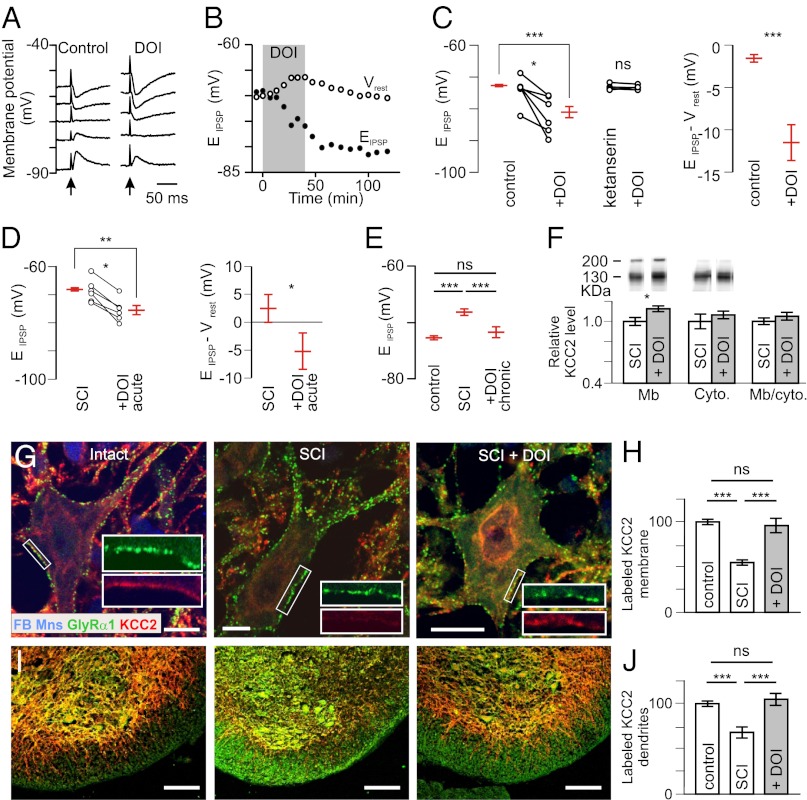
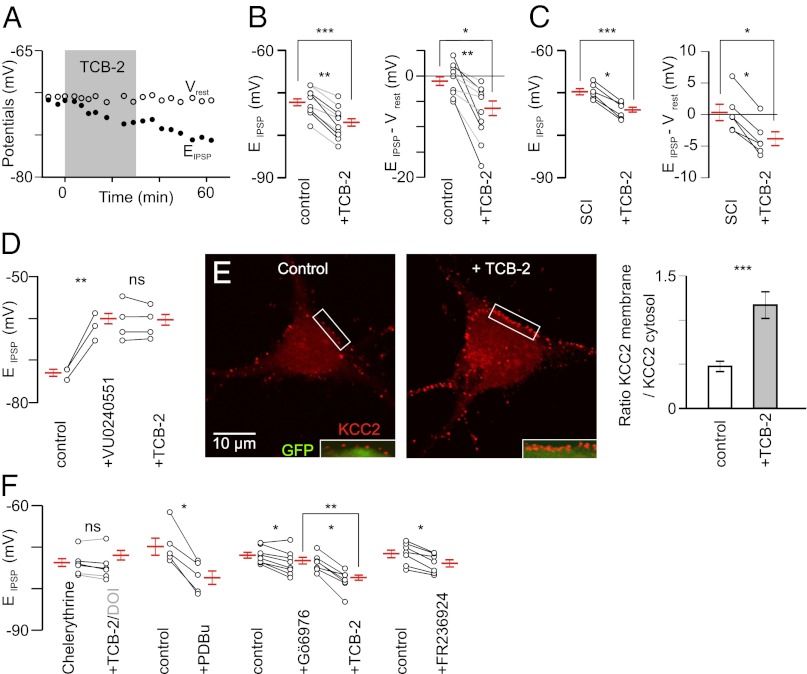
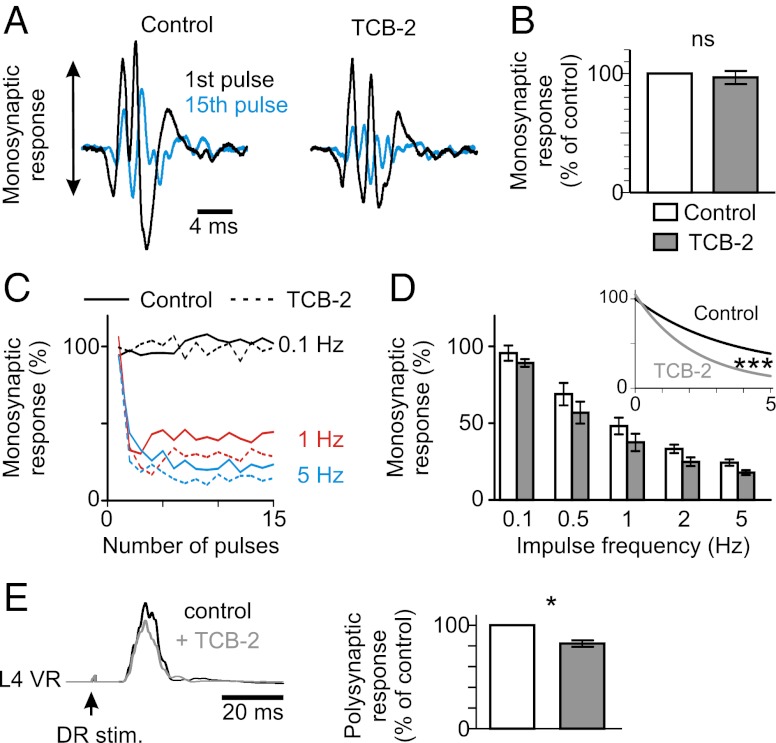
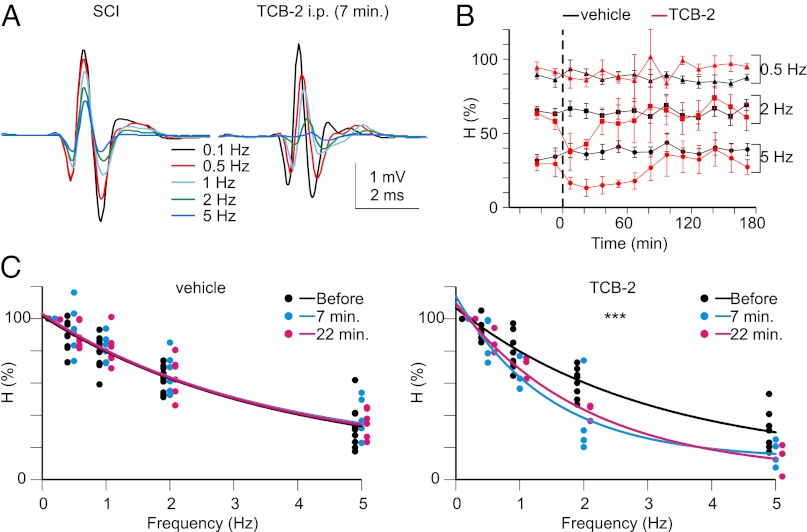
In healthy adults, activation of γ-aminobutyric acid (GABA)(A) and glycine receptors inhibits neurons as a result of low intracellular chloride concentration ([Cl(-)](i)), which is maintained by the potassium-chloride cotransporter KCC2. A reduction of KCC2 expression or function is implicated in the pathogenesis of several neurological disorders, including spasticity and chronic pain following spinal cord injury (SCI). Given the critical role of KCC2 in regulating the strength and robustness of inhibition, identifying tools that may increase KCC2 function and, hence, restore endogenous inhibition in pathological conditions is of particular importance. We show that activation of 5-hydroxytryptamine (5-HT) type 2A receptors to serotonin hyperpolarizes the reversal potential of inhibitory postsynaptic potentials (IPSPs), E(IPSP), in spinal motoneurons, increases the cell membrane expression of KCC2 and both restores endogenous inhibition and reduces spasticity after SCI in rats. Up-regulation of KCC2 function by targeting 5-HT(2A) receptors, therefore, has therapeutic potential in the treatment of neurological disorders involving altered chloride homeostasis. However, these receptors have been implicated in several psychiatric disorders, and their effects on pain processing are controversial, highlighting the need to further investigate the potential systemic effects of specific 5-HT(2A)R agonists, such as (4-bromo-3,6-dimethoxybenzocyclobuten-1-yl)methylamine hydrobromide (TCB-2).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61(6):820–838. - PubMed
-
- Payne JA, Stevenson TJ, Donaldson LF. Cation-chloride co-transporters in neuronal communication, development and trauma. J Biol Chem. 1996;271(27):16245–16252. - PubMed
-
- Payne JA, Rivera C, Voipio J, Kaila K. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. Trends Neurosci. 2003;26(4):199–206. - PubMed
-
- Kahle KT, et al. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4(9):490–503. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
