

A compound heterozygous mutation in DPAGT1 results in a congenital disorder of glycosylation with a relatively mild phenotype
- PMID: 23249953
- PMCID: PMC3722673
- DOI: 10.1038/ejhg.2012.257
A compound heterozygous mutation in DPAGT1 results in a congenital disorder of glycosylation with a relatively mild phenotype
Abstract
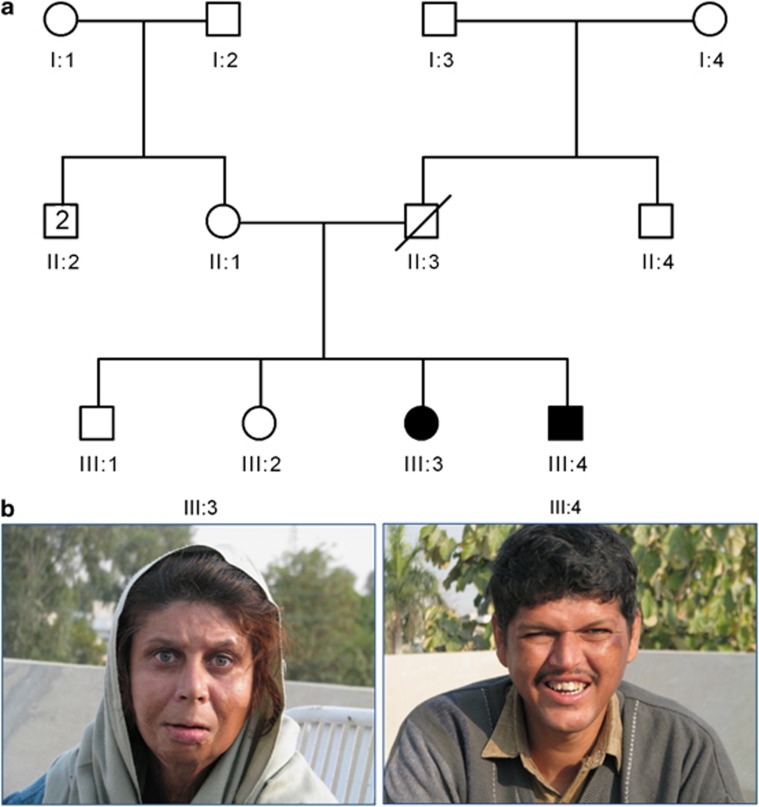
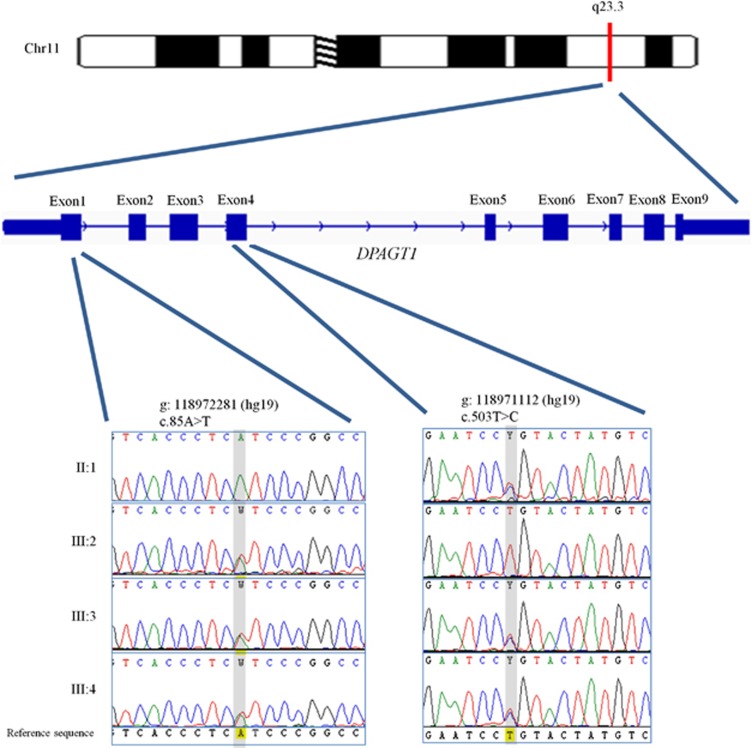
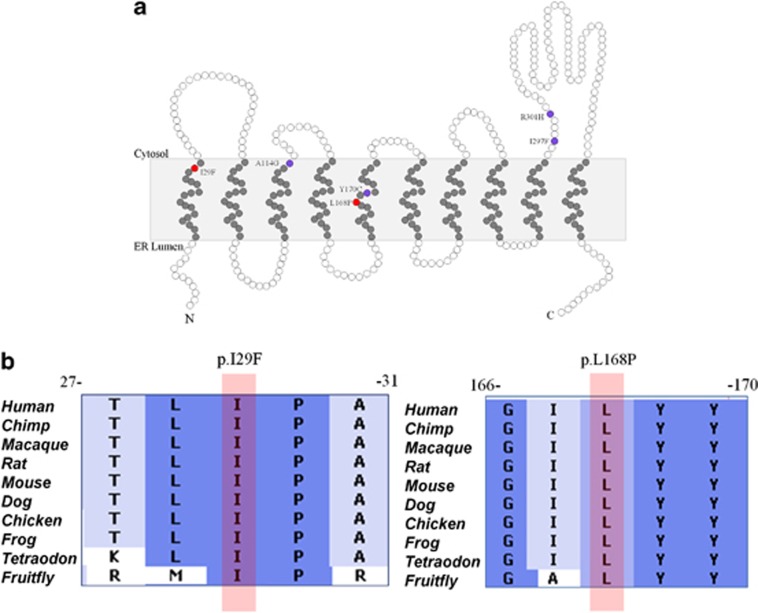
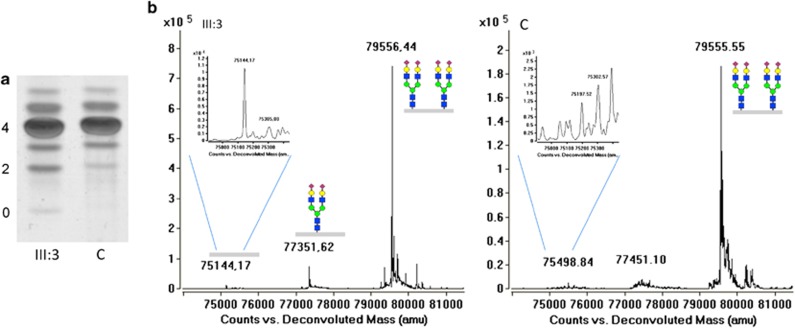
Congenital disorders of glycosylation (CDG) are a large group of recessive multisystem disorders caused by impaired protein or lipid glycosylation. The CDG-I subgroup is characterized by protein N-glycosylation defects originating in the endoplasmic reticulum. The genetic defect is known for 17 different CDG-I subtypes. Patients in the few reported DPAGT1-CDG families exhibit severe intellectual disability (ID), epilepsy, microcephaly, severe hypotonia, facial dysmorphism and structural brain anomalies. In this study, we report a non-consanguineous family with two affected adults presenting with a relatively mild phenotype consisting of moderate ID, epilepsy, hypotonia, aggressive behavior and balance problems. Exome sequencing revealed a compound heterozygous missense mutation, c.85A>T (p.I29F) and c.503T>C (p.L168P), in the DPAGT1 gene. The affected amino acids are located in the first and fifth transmembrane domains of the protein. Isoelectric focusing and high-resolution mass spectrometry analyses of serum transferrin revealed glycosylation profiles that are consistent with a CDG-I defect. Our results show that the clinical spectrum of DPAGT1-CDG is much broader than appreciated so far.
Figures
Similar articles
-
Congenital disorder of glycosylation type Ij (CDG-Ij, DPAGT1-CDG): extending the clinical and molecular spectrum of a rare disease.Mol Genet Metab. 2012 Apr;105(4):634-41. doi: 10.1016/j.ymgme.2012.01.001. Epub 2012 Jan 9. Mol Genet Metab. 2012. PMID: 22304930
-
Exome sequence identified a c.320A > G ALG13 variant in a female with infantile epileptic encephalopathy with normal glycosylation and random X inactivation: Review of the literature.Eur J Med Genet. 2017 Oct;60(10):541-547. doi: 10.1016/j.ejmg.2017.07.014. Epub 2017 Aug 1. Eur J Med Genet. 2017. PMID: 28778787 Review.
-
Fetal akinesia deformation sequence due to a congenital disorder of glycosylation.Am J Med Genet A. 2015 Oct;167A(10):2411-7. doi: 10.1002/ajmg.a.37184. Epub 2015 May 31. Am J Med Genet A. 2015. PMID: 26033833
-
DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy.Ann Neurol. 2012 Oct;72(4):550-8. doi: 10.1002/ana.23632. Ann Neurol. 2012. PMID: 23109149
-
MAN1B-CDG: Novel variants with a distinct phenotype and review of literature.Eur J Med Genet. 2019 Feb;62(2):109-114. doi: 10.1016/j.ejmg.2018.06.011. Epub 2018 Jun 14. Eur J Med Genet. 2019. PMID: 29908352 Review.
Cited by
-
Mitotic Intragenic Recombination: A Mechanism of Survival for Several Congenital Disorders of Glycosylation.Am J Hum Genet. 2016 Feb 4;98(2):339-46. doi: 10.1016/j.ajhg.2015.12.007. Epub 2016 Jan 21. Am J Hum Genet. 2016. PMID: 26805780 Free PMC article.
-
Structures of DPAGT1 Explain Glycosylation Disease Mechanisms and Advance TB Antibiotic Design.Cell. 2018 Nov 1;175(4):1045-1058.e16. doi: 10.1016/j.cell.2018.10.037. Cell. 2018. PMID: 30388443 Free PMC article.
-
A missense mutation in a patient with developmental delay affects the activity and structure of the hexosamine biosynthetic pathway enzyme AGX1.FEBS Lett. 2021 Jan;595(1):110-122. doi: 10.1002/1873-3468.13968. Epub 2020 Nov 18. FEBS Lett. 2021. PMID: 33098688 Free PMC article.
-
Glycosylation and behavioral symptoms in neurological disorders.Transl Psychiatry. 2023 May 8;13(1):154. doi: 10.1038/s41398-023-02446-x. Transl Psychiatry. 2023. PMID: 37156804 Free PMC article. Review.
-
Clinical and genetic characterisation of a large Indian congenital myasthenic syndrome cohort.Brain. 2024 Jan 4;147(1):281-296. doi: 10.1093/brain/awad315. Brain. 2024. PMID: 37721175 Free PMC article.
References
-
- Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–198. - PubMed
-
- Theodore M, Morava E. Congenital disorders of glycosylation: sweet news. Curr Opin Pediatr. 2011;23:581–587. - PubMed
-
- Wu X, Rush JS, Karaoglu D, et al. Deficiency of UDP-GlcNAc:dolichol phosphate n-acetylglucosamine-1 phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum Mutat. 2003;22:144–150. - PubMed
-
- Freeze HH. Human disorders in N-glycosylation and animal models. Biochim Biophys Acta. 2002;1573:388–393. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
