

Expression of mutant huntingtin in leptin receptor-expressing neurons does not control the metabolic and psychiatric phenotype of the BACHD mouse
- PMID: 23251447
- PMCID: PMC3519539
- DOI: 10.1371/journal.pone.0051168
Expression of mutant huntingtin in leptin receptor-expressing neurons does not control the metabolic and psychiatric phenotype of the BACHD mouse
Abstract
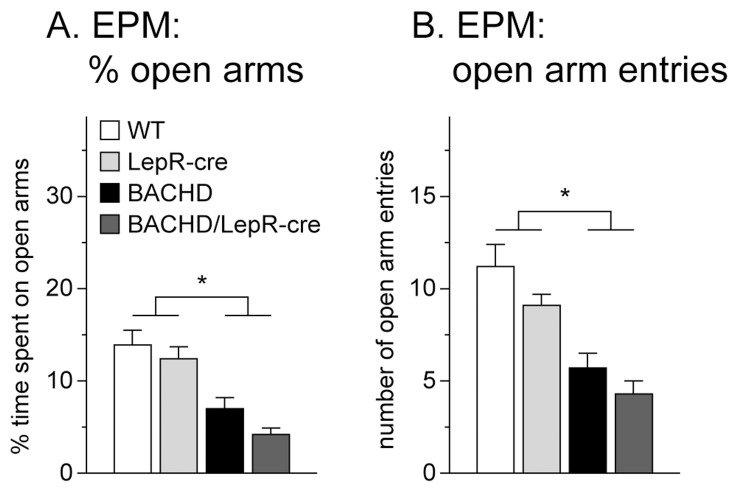
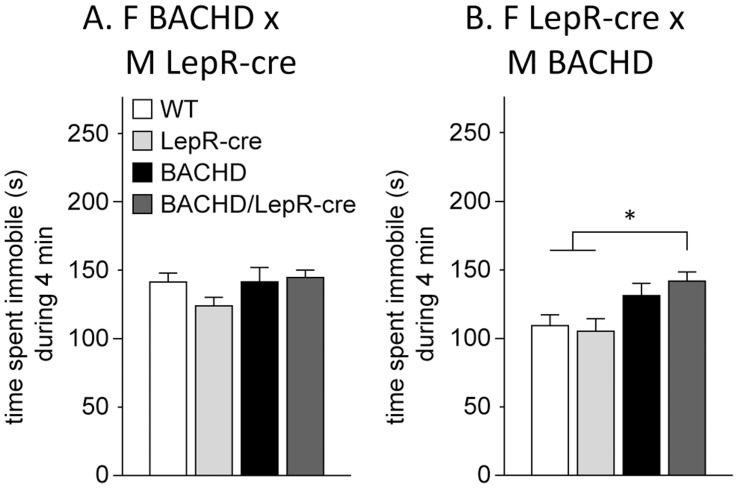
Metabolic and psychiatric disturbances occur early on in the clinical manifestation of Huntington's disease (HD), a neurodegenerative disorder caused by an expanded CAG repeat in the huntingtin (HTT) gene. Hypothalamus has emerged as an important site of pathology and alterations in this area and its neuroendocrine circuits may play a role in causing early non-motor symptoms and signs in HD. Leptin is a hormone that controls energy homeostasis by signaling through leptin receptors in the hypothalamus. Disturbed leptin action is implicated in both obesity and depression and altered circulating levels of leptin have been reported in both clinical HD and rodent models of the disease. Pathological leptin signaling may therefore be involved in causing the metabolic and psychiatric disturbances of HD. Here we tested the hypothesis that expression of mutant HTT in leptin receptor carrying neurons plays a role in the development of the non-motor phenotype in the BACHD mouse model. Our results show that inactivation of mutant HTT in leptin receptor-expressing neurons in the BACHD mouse using cross-breeding based on a cre-loxP system did not have an effect on the metabolic phenotype or anxiety-like behavior. The data suggest that mutant HTT disrupts critical hypothalamic pathways by other mechanisms than interfering with intracellular leptin signaling.
Conflict of interest statement
Figures
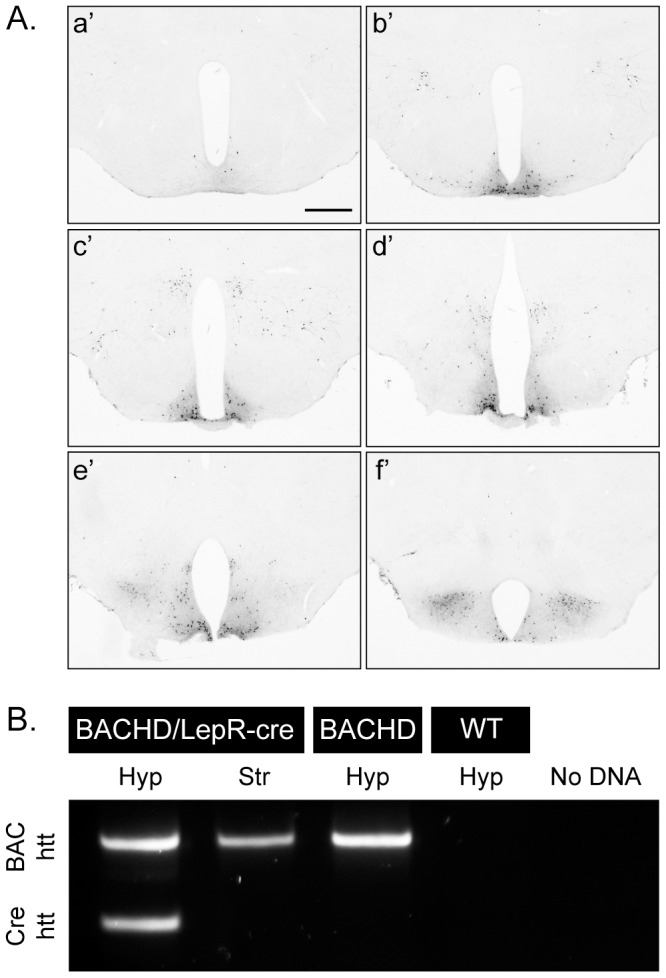
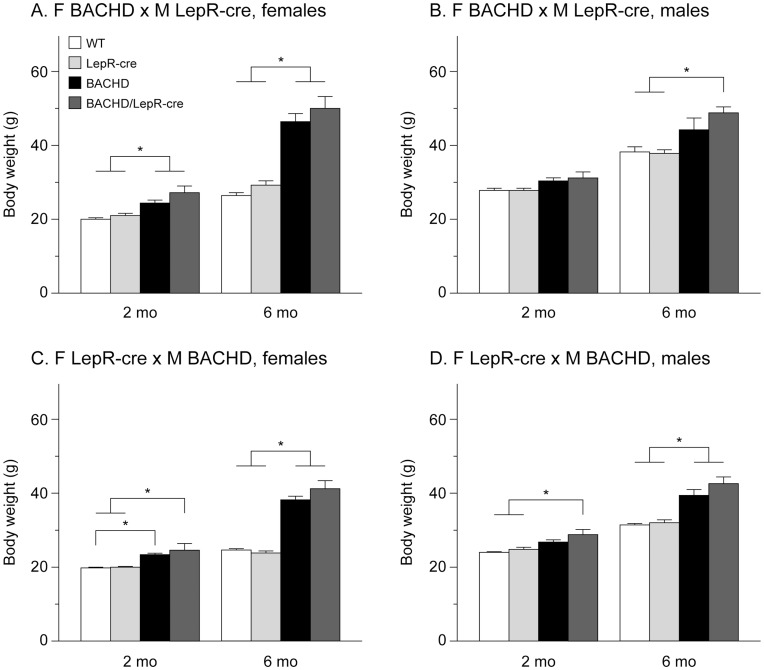
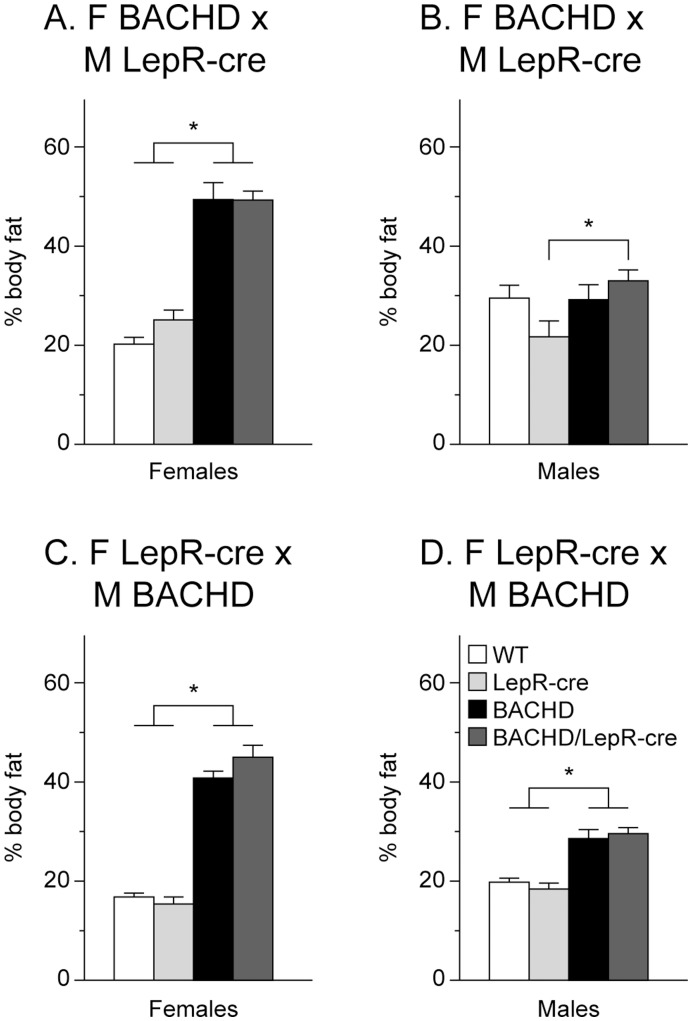
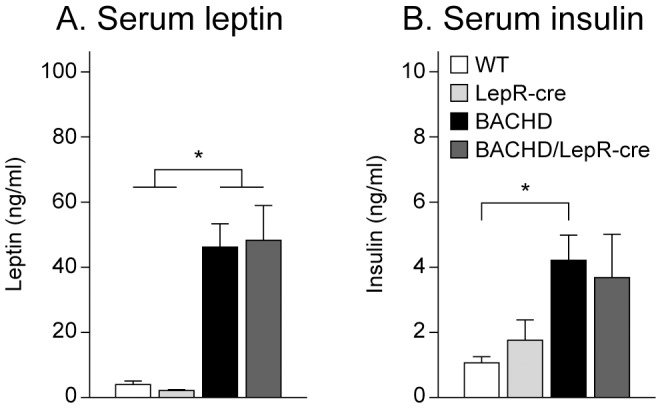
References
-
- HDCRG (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72: 971–983. - PubMed
-
- Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10: 83–98. - PubMed
-
- Sturrock A, Leavitt BR (2010) The clinical and genetic features of Huntington disease. J Geriatr Psychiatry Neurol 23: 243–259. - PubMed
-
- Aziz NA, Swaab DF, Pijl H, Roos RA (2007) Hypothalamic dysfunction and neuroendocrine and metabolic alterations in Huntington’s disease: clinical consequences and therapeutic implications. Rev Neurosci 18: 223–251. - PubMed
-
- Lalic NM, Maric J, Svetel M, Jotic A, Stefanova E, et al. (2008) Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol 65: 476–480. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
