

Matrices of physiologic stiffness potently inactivate idiopathic pulmonary fibrosis fibroblasts
- PMID: 23258227
- PMCID: PMC3653602
- DOI: 10.1165/rcmb.2012-0335OC
Matrices of physiologic stiffness potently inactivate idiopathic pulmonary fibrosis fibroblasts
Abstract
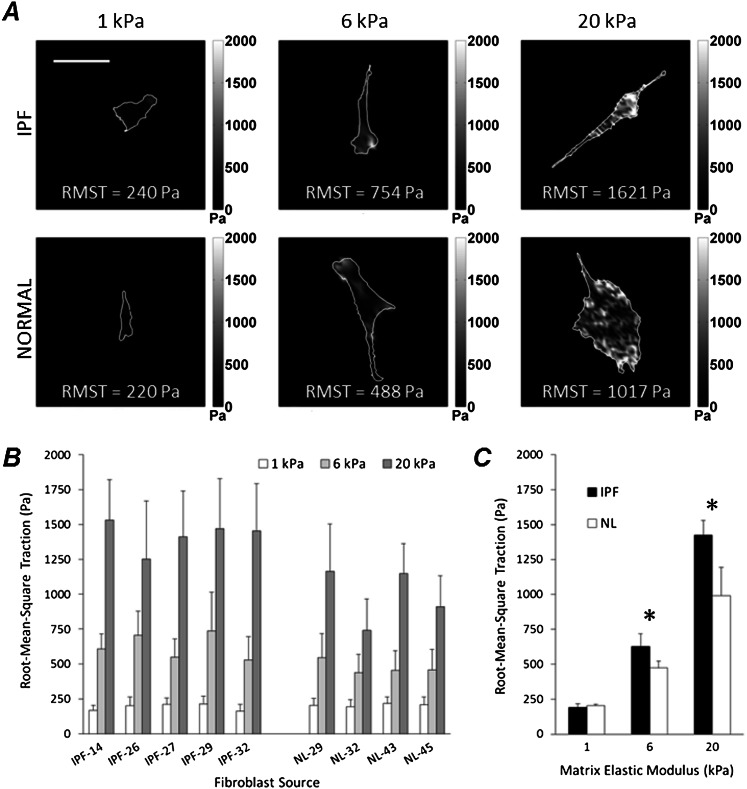
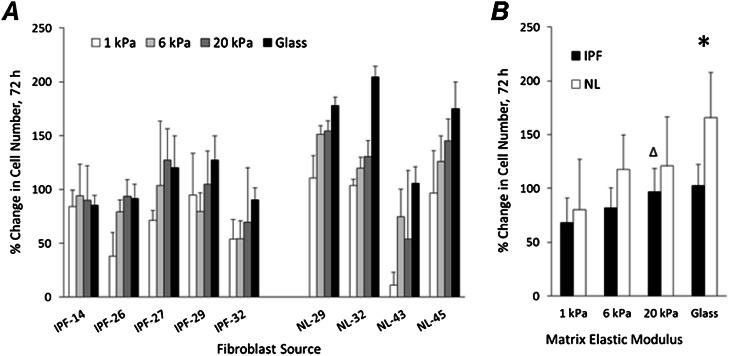
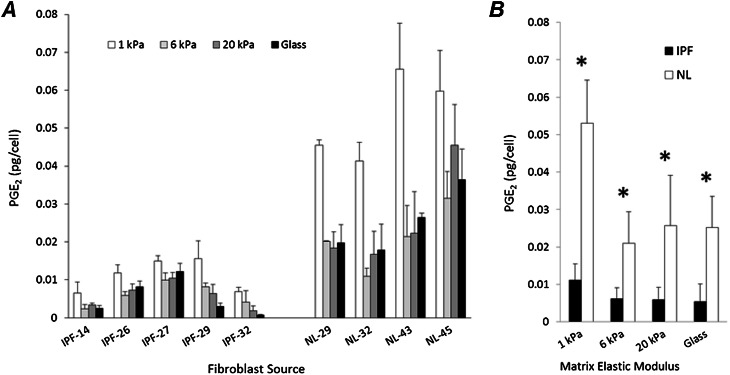
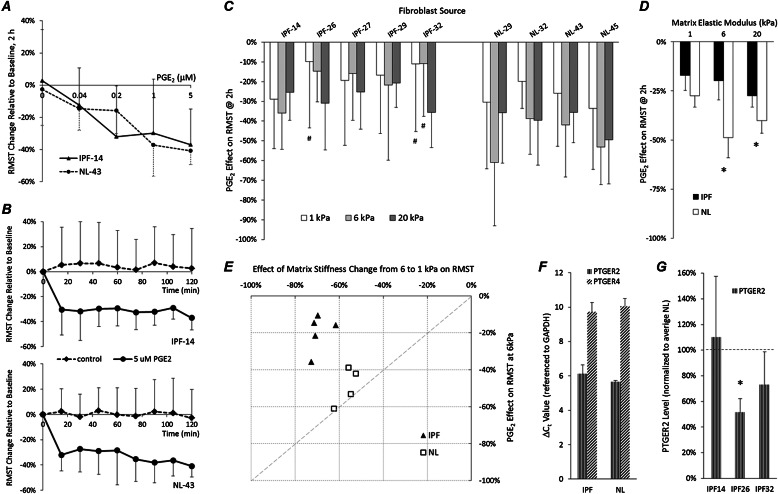
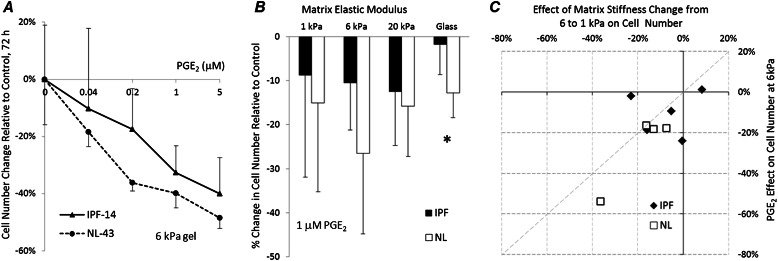
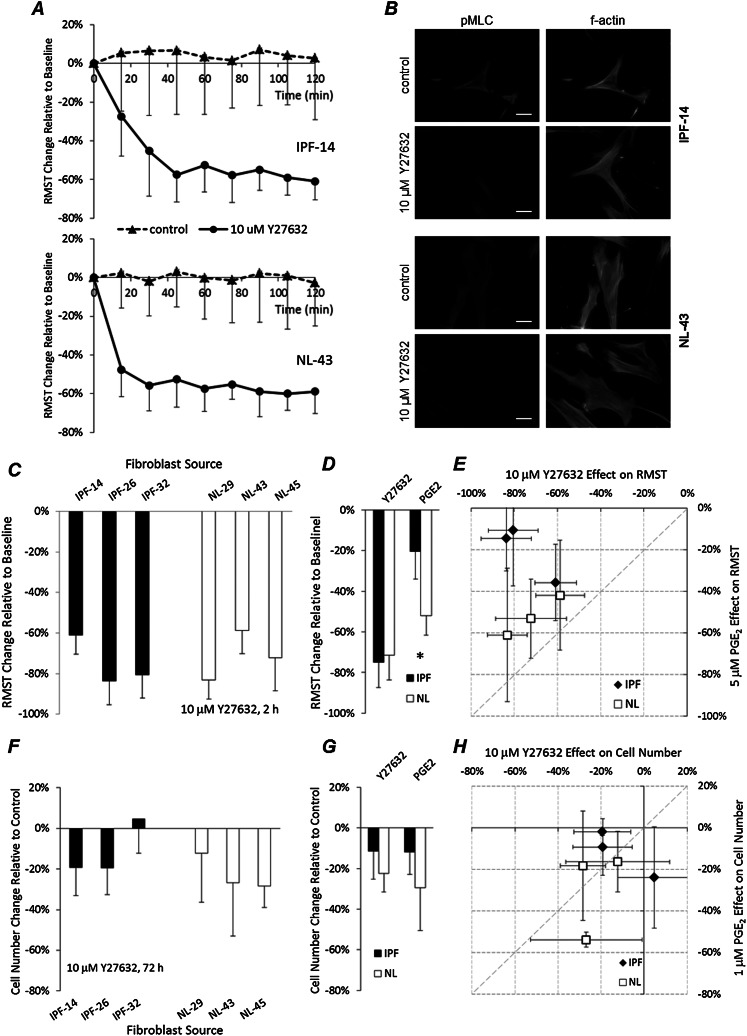
Fibroblasts from patients with idiopathic pulmonary fibrosis (IPF) have been shown to differ from normal lung fibroblasts in functional behaviors that contribute to the pathogenesis of IPF, including the expression of contractile proteins and proliferation, but how such behaviors vary in matrices with stiffness matched to normal and fibrotic lung tissue remains unknown. Here, we tested whether pathologic changes in matrix stiffness control IPF and normal lung tissue-derived fibroblast functions, and compared the relative efficacy of mechanical cues to an antifibrotic lipid mediator, prostaglandin E(2) (PGE(2)). Fibroblasts were grown on collagen I-coated glass or hydrogel substrates of discrete stiffnesses, spanning the range of normal and fibrotic lung tissue. Traction microscopy was used to quantify contractile function. The CyQuant Cell Proliferation Assay (Invitrogen, Carlsbad, CA) was used to assess changes in cell number, and PGE(2) concentrations were measured by ELISA. We confirmed differences in proliferation and PGE(2) synthesis between IPF and normal tissue-derived fibroblasts on rigid substrates. However, IPF fibroblasts remained highly responsive to changes in matrix stiffness, and both proliferative and contractile differences between IPF and normal fibroblasts were ablated on physiologically soft matrices. We also confirmed the relative resistance of IPF fibroblasts to PGE(2), while demonstrating that decreases in matrix stiffness and the inhibition of Rho kinase both potently attenuate contractile function in IPF-derived fibroblasts. We conclude that pathologic changes in the mechanical environment control important IPF fibroblast functions. Understanding how mechanical cues control fibroblast function may offer new opportunities for targeting these cells, even when they are resistant to antifibrotic pharmacological agents or biological mediators.
Figures
Similar articles
-
Extracellular Matrix Cross-Linking Enhances Fibroblast Growth and Protects against Matrix Proteolysis in Lung Fibrosis.Am J Respir Cell Mol Biol. 2018 May;58(5):594-603. doi: 10.1165/rcmb.2016-0379OC. Am J Respir Cell Mol Biol. 2018. PMID: 29053339
-
Fibroblast-matrix interplay: Nintedanib and pirfenidone modulate the effect of IPF fibroblast-conditioned matrix on normal fibroblast phenotype.Respirology. 2018 Aug;23(8):756-763. doi: 10.1111/resp.13287. Epub 2018 Mar 12. Respirology. 2018. PMID: 29532550
-
SEMA3B inhibits TGFβ-induced extracellular matrix protein production and its reduced levels are associated with a decline in lung function in IPF.Am J Physiol Cell Physiol. 2024 Jun 1;326(6):C1659-C1668. doi: 10.1152/ajpcell.00681.2023. Epub 2024 Apr 22. Am J Physiol Cell Physiol. 2024. PMID: 38646784 Free PMC article.
-
Lung Fibroblasts, Aging, and Idiopathic Pulmonary Fibrosis.Ann Am Thorac Soc. 2016 Dec;13 Suppl 5:S417-S421. doi: 10.1513/AnnalsATS.201605-341AW. Ann Am Thorac Soc. 2016. PMID: 28005427 Review.
-
3D in vitro hydrogel models to study the human lung extracellular matrix and fibroblast function.Respir Res. 2023 Oct 5;24(1):242. doi: 10.1186/s12931-023-02548-6. Respir Res. 2023. PMID: 37798767 Free PMC article. Review.
Cited by
-
Second harmonic generation microscopy reveals altered collagen microstructure in usual interstitial pneumonia versus healthy lung.Respir Res. 2015 May 27;16(1):61. doi: 10.1186/s12931-015-0220-8. Respir Res. 2015. PMID: 26013144 Free PMC article.
-
Matrix metalloproteinase-7 is increased in lung bases but not apices in idiopathic pulmonary fibrosis.ERJ Open Res. 2022 Oct 24;8(4):00191-2022. doi: 10.1183/23120541.00191-2022. eCollection 2022 Oct. ERJ Open Res. 2022. PMID: 36299365 Free PMC article.
-
Matrix Mechanics as Regulatory Factors and Therapeutic Targets in Hepatic Fibrosis.Int J Biol Sci. 2019 Sep 7;15(12):2509-2521. doi: 10.7150/ijbs.37500. eCollection 2019. Int J Biol Sci. 2019. PMID: 31754325 Free PMC article. Review.
-
Epithelial-stromal cell interactions and extracellular matrix mechanics drive the formation of airway-mimetic tubular morphology in lung organoids.iScience. 2021 Aug 30;24(9):103061. doi: 10.1016/j.isci.2021.103061. eCollection 2021 Sep 24. iScience. 2021. PMID: 34585112 Free PMC article.
-
Exploring therapeutic targets for molecular therapy of idiopathic pulmonary fibrosis.Sci Prog. 2024 Apr-Jun;107(2):368504241247402. doi: 10.1177/00368504241247402. Sci Prog. 2024. PMID: 38651330 Free PMC article. Review.
References
-
- Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998;157:1301–1315 - PubMed
-
- American Thoracic Society and European Respiratory Society Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. Am J Respir Crit Care Med 2000;161:646–664 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
