

Rotenone inhibits autophagic flux prior to inducing cell death
- PMID: 23259041
- PMCID: PMC3526971
- DOI: 10.1021/cn300145z
Rotenone inhibits autophagic flux prior to inducing cell death
Abstract
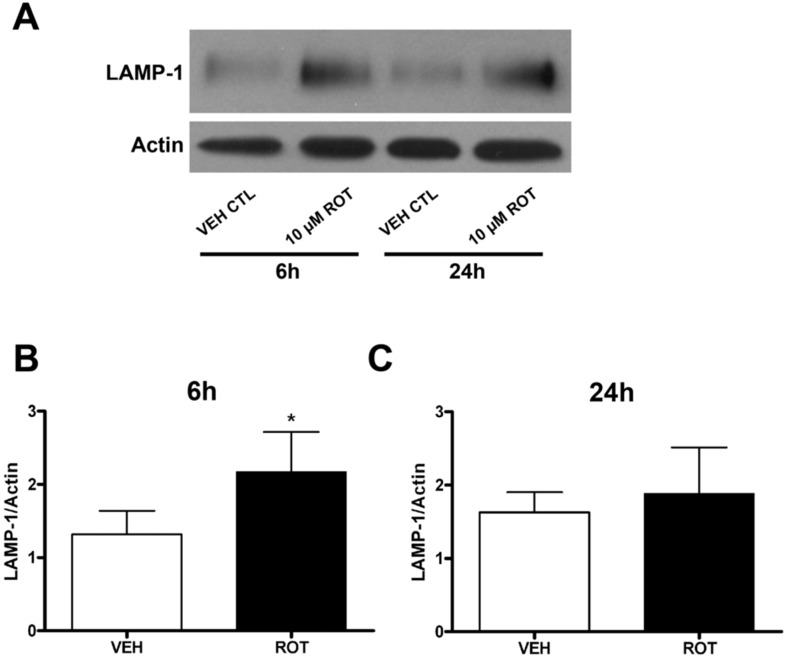
Rotenone, which selectively inhibits mitochondrial complex I, induces oxidative stress, α-synuclein accumulation, and dopaminergic neuron death, principal pathological features of Parkinson's disease. The autophagy-lysosome pathway degrades damaged proteins and organelles for the intracellular maintenance of nutrient and energy balance. While it is known that rotenone causes autophagic vacuole accumulation, the mechanism by which this effect occurs has not been thoroughly investigated. Treatment of differentiated SH-SY5Y cells with rotenone (10 μM) induced the accumulation of autophagic vacuoles at 6 h and 24 h as indicated by Western blot analysis for microtubule associated protein-light chain 3-II (MAP-LC3-II). Assessment of autophagic flux at these time points indicated that autophagic vacuole accumulation resulted from a decrease in their effective lysosomal degradation, which was substantiated by increased levels of autophagy substrates p62 and α-synuclein. Inhibition of lysosomal degradation may be explained by the observed decrease in cellular ATP levels, which in turn may have caused the observed concomitant increase in acidic vesicle pH. The early (6 h) effects of rotenone on cellular energetics and autophagy-lysosome pathway function preceded the induction of cell death and apoptosis. These findings indicate that the classical mitochondrial toxin rotenone has a pronounced effect on macroautophagy completion that may contribute to its neurotoxic potential.
Figures






References
-
- Olanow C. W.; Stern M. B.; Sethi K. (2009) The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology 72, S1–136. - PubMed
-
- Dauer W.; Przedborski S. (2003) Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. - PubMed
-
- Klionsky D. J.; Abeliovich H.; Agostinis P.; Agrawal D. K.; Aliev G.; Askew D. S.; Baba M.; Baehrecke E. H.; Bahr B. A.; Ballabio A.; Bamber B. A.; Bassham D. C.; Bergamini E.; Bi X.; Biard-Piechaczyk M.; Blum J. S.; Bredesen D. E.; Brodsky J. L.; Brumell J. H.; Brunk U. T.; Bursch W.; Camougrand N.; Cebollero E.; Cecconi F.; Chen Y.; Chin L. S.; Choi A.; Chu C. T.; Chung J.; Clarke P. G.; Clark R. S.; Clarke S. G.; Clave C.; Cleveland J. L.; Codogno P.; Colombo M. I.; Coto-Montes A.; Cregg J. M.; Cuervo A. M.; Debnath J.; Demarchi F.; Dennis P. B.; Dennis P. A.; Deretic V.; Devenish R. J.; Di Sano F.; Dice J. F.; Difiglia M.; Dinesh-Kumar S.; Distelhorst C. W.; Djavaheri-Mergny M.; Dorsey F. C.; Droge W.; Dron M.; Dunn W. A. Jr.; Duszenko M.; Eissa N. T.; Elazar Z.; Esclatine A.; Eskelinen E. L.; Fesus L.; Finley K. D.; Fuentes J. M.; Fueyo J.; Fujisaki K.; Galliot B.; Gao F. B.; Gewirtz D. A.; Gibson S. B.; Gohla A.; Goldberg A. L.; Gonzalez R.; Gonzalez-Estevez C.; Gorski S.; Gottlieb R. A.; Haussinger D.; He Y. W.; Heidenreich K.; Hill J. A.; Hoyer-Hansen M.; Hu X.; Huang W. P.; Iwasaki A.; Jaattela M.; Jackson W. T.; Jiang X.; Jin S.; Johansen T.; Jung J. U.; Kadowaki M.; Kang C.; Kelekar A.; Kessel D. H.; Kiel J. A.; Kim H. P.; Kimchi A.; Kinsella T. J.; Kiselyov K.; Kitamoto K.; Knecht E.; Komatsu M.; Kominami E.; Kondo S.; Kovacs A. L.; Kroemer G.; Kuan C. Y.; Kumar R.; Kundu M.; Landry J.; Laporte M.; Le W.; Lei H. Y.; Lenardo M. J.; Levine B.; Lieberman A.; Lim K. L.; Lin F. C.; Liou W.; Liu L. F.; Lopez-Berestein G.; Lopez-Otin C.; Lu B.; Macleod K. F.; Malorni W.; Martinet W.; Matsuoka K.; Mautner J.; Meijer A. J.; Melendez A.; Michels P.; Miotto G.; Mistiaen W. P.; Mizushima N.; Mograbi B.; Monastyrska I.; Moore M. N.; Moreira P. I.; Moriyasu Y.; Motyl T.; Munz C.; Murphy L. O.; Naqvi N. I.; Neufeld T. P.; Nishino I.; Nixon R. A.; Noda T.; Nurnberg B.; Ogawa M.; Oleinick N. L.; Olsen L. J.; Ozpolat B.; Paglin S.; Palmer G. E.; Papassideri I.; Parkes M.; Perlmutter D. H.; Perry G.; Piacentini M.; Pinkas-Kramarski R.; Prescott M.; Proikas-Cezanne T.; Raben N.; Rami A.; Reggiori F.; Rohrer B.; Rubinsztein D. C.; Ryan K. M.; Sadoshima J.; Sakagami H.; Sakai Y.; Sandri M.; Sasakawa C.; Sass M.; Schneider C.; Seglen P. O.; Seleverstov O.; Settleman J.; Shacka J. J.; Shapiro I. M.; Sibirny A.; Silva-Zacarin E. C.; Simon H. U.; Simone C.; Simonsen A.; Smith M. A.; Spanel-Borowski K.; Srinivas V.; Steeves M.; Stenmark H.; Stromhaug P. E.; Subauste C. S.; Sugimoto S.; Sulzer D.; Suzuki T.; Swanson M. S.; Tabas I.; Takeshita F.; Talbot N. J.; Talloczy Z.; Tanaka K.; Tanida I.; Taylor G. S.; Taylor J. P.; Terman A.; Tettamanti G.; Thompson C. B.; Thumm M.; Tolkovsky A. M.; Tooze S. A.; Truant R.; Tumanovska L. V.; Uchiyama Y.; Ueno T.; Uzcategui N. L.; van der Klei I.; Vaquero E. C.; Vellai T.; Vogel M. W.; Wang H. G.; Webster P.; Wiley J. W.; Xi Z.; Xiao G.; Yahalom J.; Yang J. M.; Yap G.; Yin X. M.; Yoshimori T.; Yu L.; Yue Z.; Yuzaki M.; Zabirnyk O.; Zheng X.; Zhu X.; Deter R. L. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175. - PMC - PubMed
-
- Shacka J. J.; Roth K. A.; Zhang J. (2008) The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front. Biosci. 13, 718–736. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
