

Ray Meta: scalable de novo metagenome assembly and profiling
- PMID: 23259615
- PMCID: PMC4056372
- DOI: 10.1186/gb-2012-13-12-r122
Ray Meta: scalable de novo metagenome assembly and profiling
Abstract
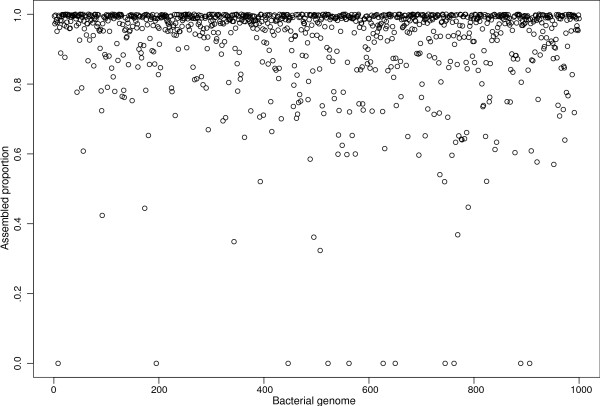
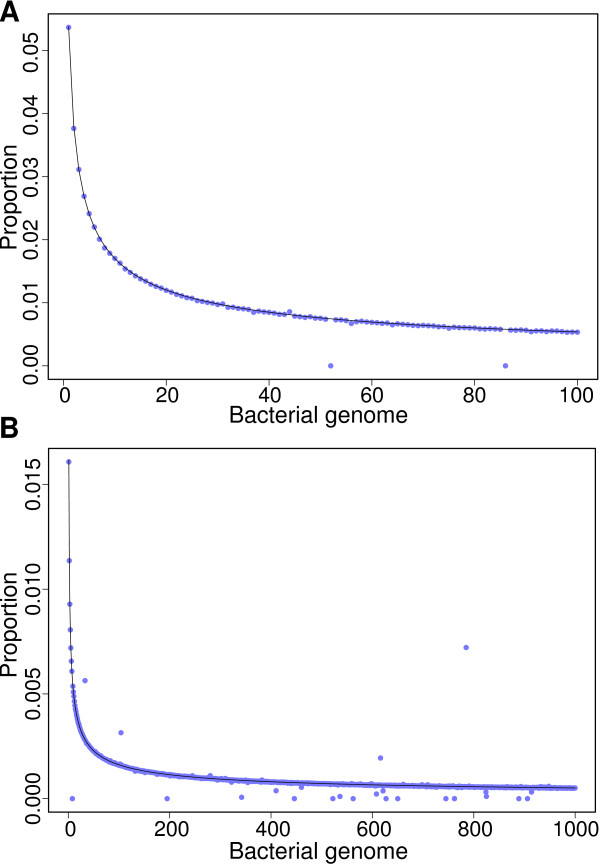
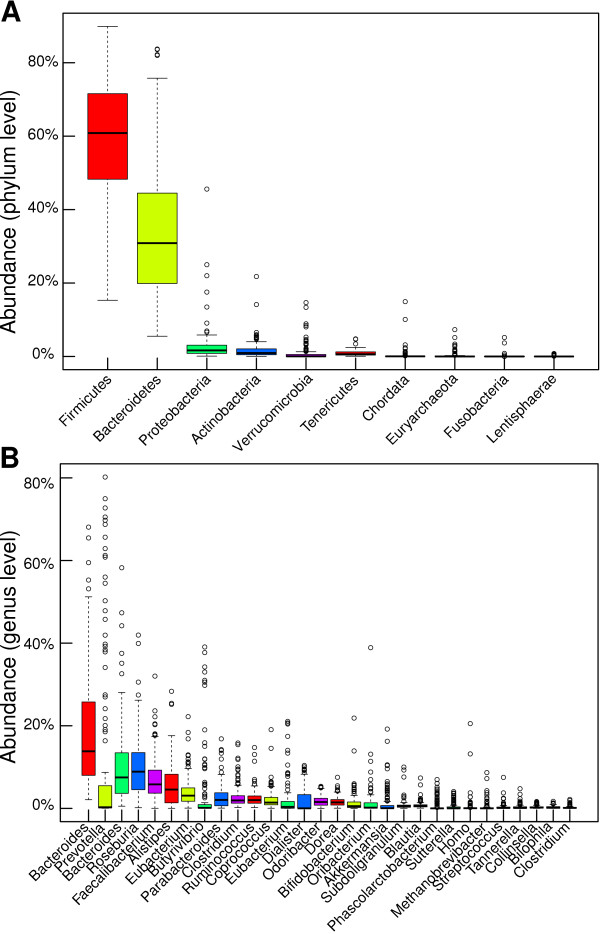
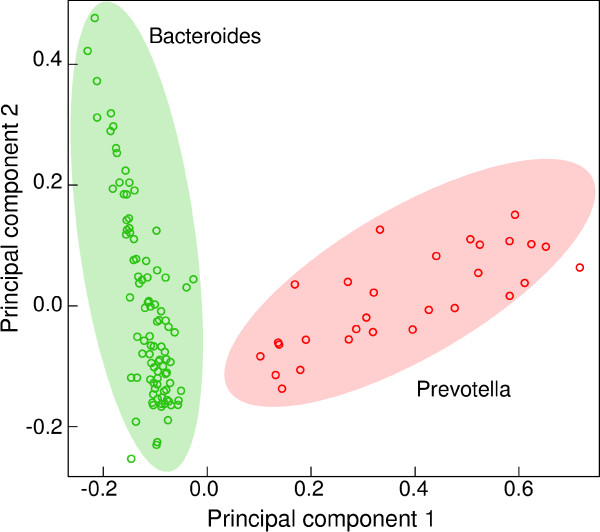
Voluminous parallel sequencing datasets, especially metagenomic experiments, require distributed computing for de novo assembly and taxonomic profiling. Ray Meta is a massively distributed metagenome assembler that is coupled with Ray Communities, which profiles microbiomes based on uniquely-colored k-mers. It can accurately assemble and profile a three billion read metagenomic experiment representing 1,000 bacterial genomes of uneven proportions in 15 hours with 1,024 processor cores, using only 1.5 GB per core. The software will facilitate the processing of large and complex datasets, and will help in generating biological insights for specific environments. Ray Meta is open source and available at http://denovoassembler.sf.net.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
