

Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights
- PMID: 23264687
- PMCID: PMC3786661
- DOI: 10.1136/jnnp-2012-304019
Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights
Abstract
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disorder of the motor neurons in the motor cortex, brainstem and spinal cord. A combination of upper and lower motor neuron dysfunction comprises the clinical ALS phenotype. Although the ALS phenotype was first observed by Charcot over 100 years ago, the site of ALS onset and the pathophysiological mechanisms underlying the development of motor neuron degeneration remain to be elucidated. Transcranial magnetic stimulation (TMS) enables non-invasive assessment of the functional integrity of the motor cortex and its corticomotoneuronal projections. To date, TMS studies have established motor cortical and corticospinal dysfunction in ALS, with cortical hyperexcitability being an early feature in sporadic forms of ALS and preceding the clinical onset of familial ALS. Taken together, a central origin of ALS is supported by TMS studies, with an anterograde transsynaptic mechanism implicated in ALS pathogenesis. Of further relevance, TMS techniques reliably distinguish ALS from mimic disorders, despite a compatible peripheral disease burden, thereby suggesting a potential diagnostic utility of TMS in ALS. This review will focus on the mechanisms underlying the generation of TMS measures used in assessment of cortical excitability, the contribution of TMS in enhancing the understanding of ALS pathophysiology and the potential diagnostic utility of TMS techniques in ALS.
Keywords: ALS; EMG.
Figures
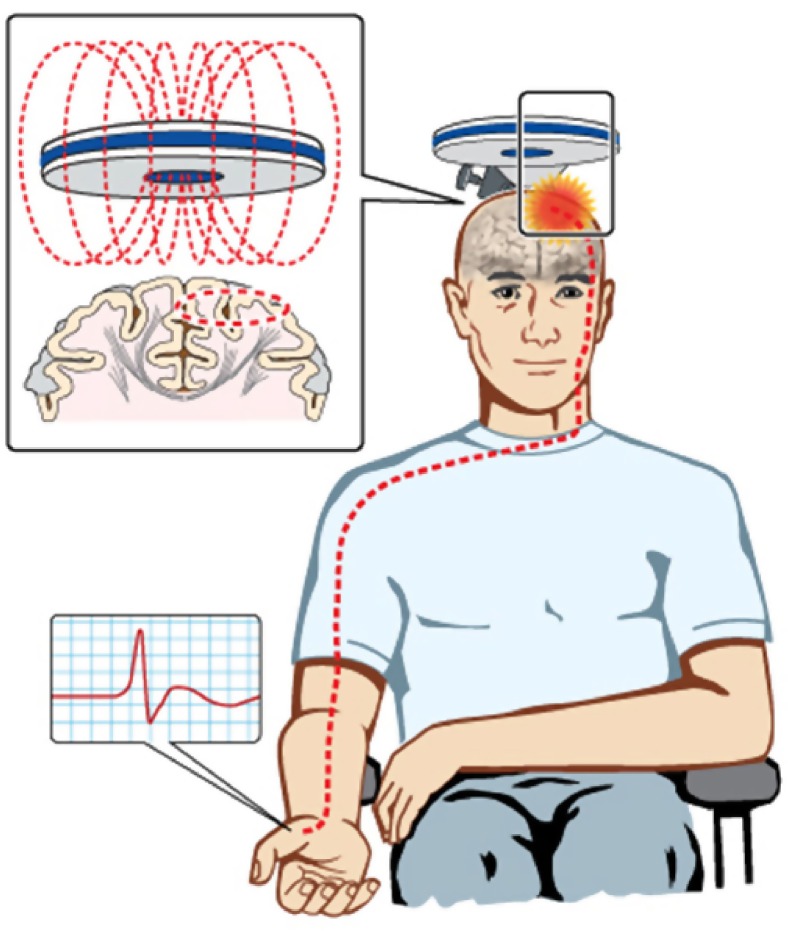
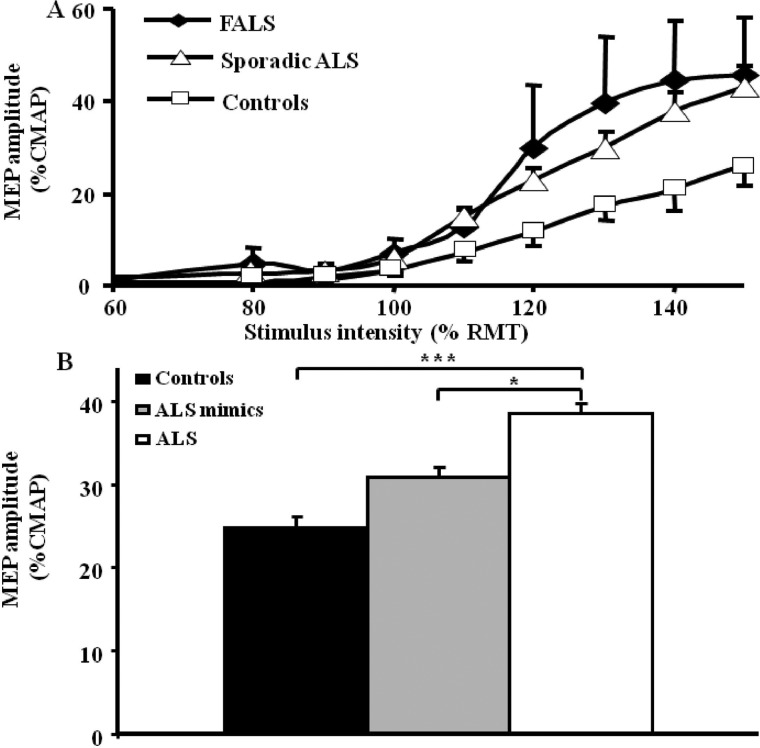
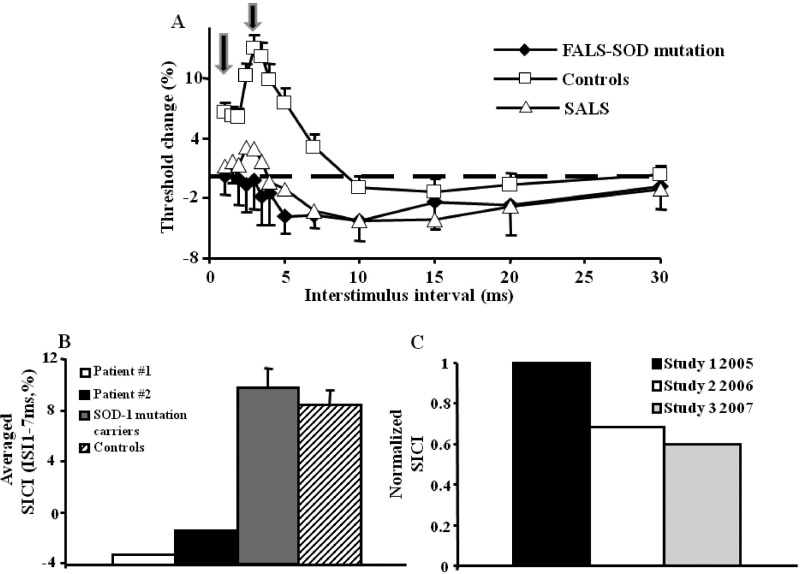
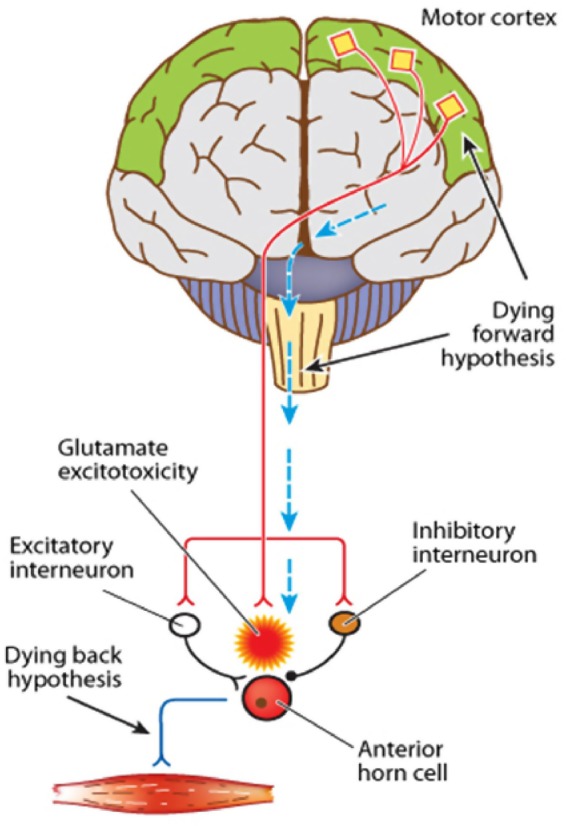
References
-
- Charcot J, Joffroy A. Deux cas d'atrophie musculaire progressive avec lesion de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch Physiol Neurol Pathol 1869;2:744–54
-
- Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–55 - PubMed
-
- Vucic S, Burke D, Kiernan MC. Diagnosis of motor neuron disease. In: Kiernan MC. ed. The motor neuron disease handbook. Sydney: Australasian Medical Publishing Company Limited, 2007:89–115
-
- Winhammar JM, Rowe DB, Henderson RD, et al. Assessment of disease progression in motor neuron disease. Lancet Neurology 2005;4:229–38 - PubMed
-
- Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007;68:1571–5 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous