

The variance of identity-by-descent sharing in the Wright-Fisher model
- PMID: 23267057
- PMCID: PMC3584006
- DOI: 10.1534/genetics.112.147215
The variance of identity-by-descent sharing in the Wright-Fisher model
Abstract
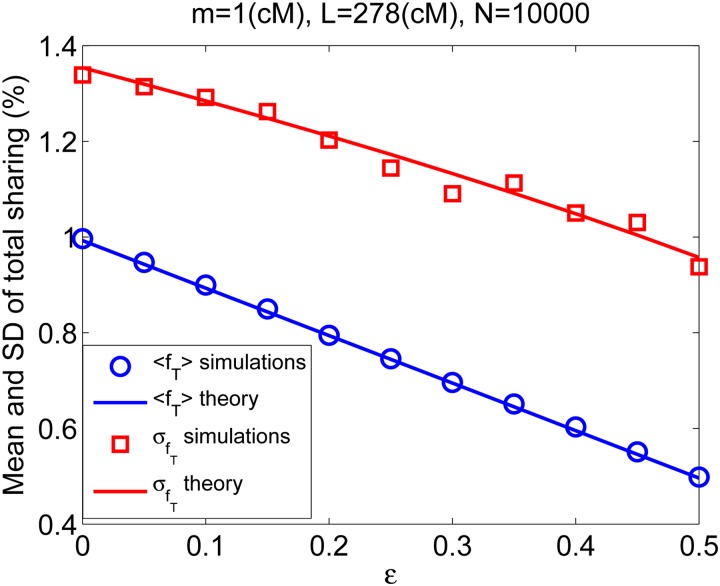
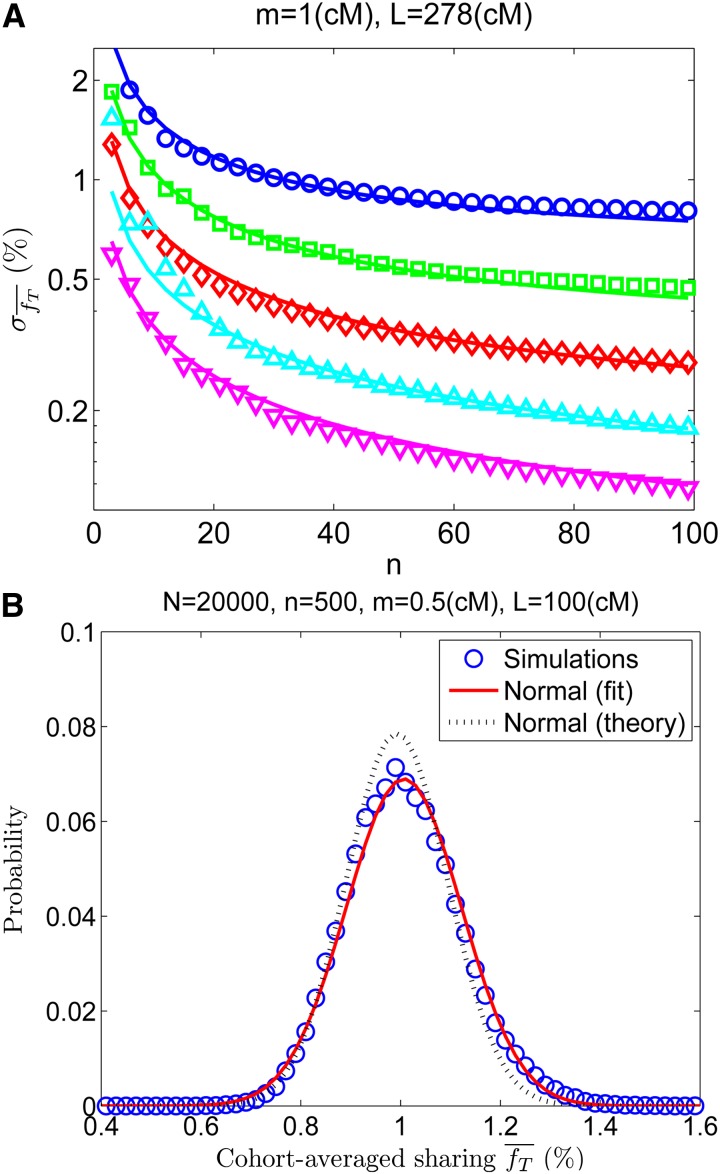
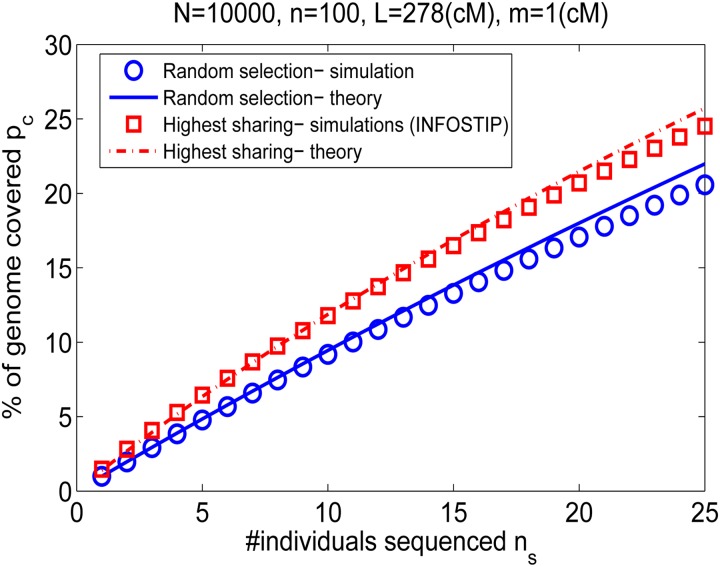
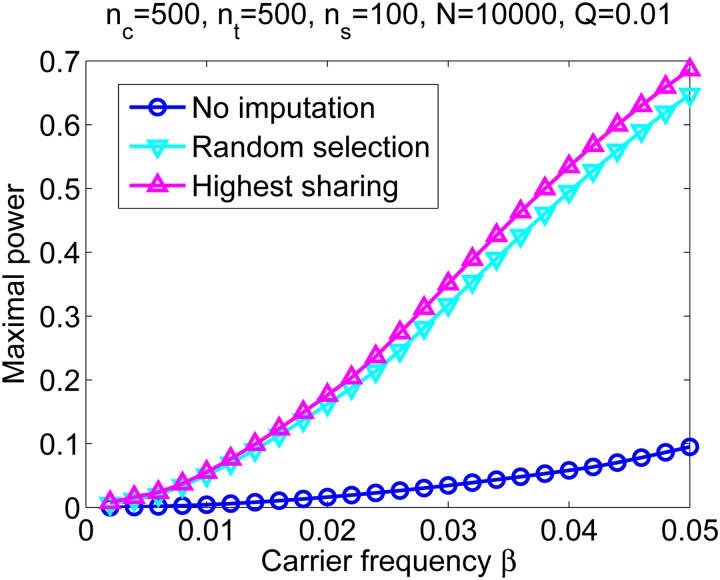
Widespread sharing of long, identical-by-descent (IBD) genetic segments is a hallmark of populations that have experienced recent genetic drift. Detection of these IBD segments has recently become feasible, enabling a wide range of applications from phasing and imputation to demographic inference. Here, we study the distribution of IBD sharing in the Wright-Fisher model. Specifically, using coalescent theory, we calculate the variance of the total sharing between random pairs of individuals. We then investigate the cohort-averaged sharing: the average total sharing between one individual and the rest of the cohort. We find that for large cohorts, the cohort-averaged sharing is distributed approximately normally. Surprisingly, the variance of this distribution does not vanish even for large cohorts, implying the existence of "hypersharing" individuals. The presence of such individuals has consequences for the design of sequencing studies, since, if they are selected for whole-genome sequencing, a larger fraction of the cohort can be subsequently imputed. We calculate the expected gain in power of imputation by IBD and subsequently in power to detect an association, when individuals are either randomly selected or specifically chosen to be the hypersharing individuals. Using our framework, we also compute the variance of an estimator of the population size that is based on the mean IBD sharing and the variance in the sharing between inbred siblings. Finally, we study IBD sharing in an admixture pulse model and show that in the Ashkenazi Jewish population the admixture fraction is correlated with the cohort-averaged sharing.
Figures
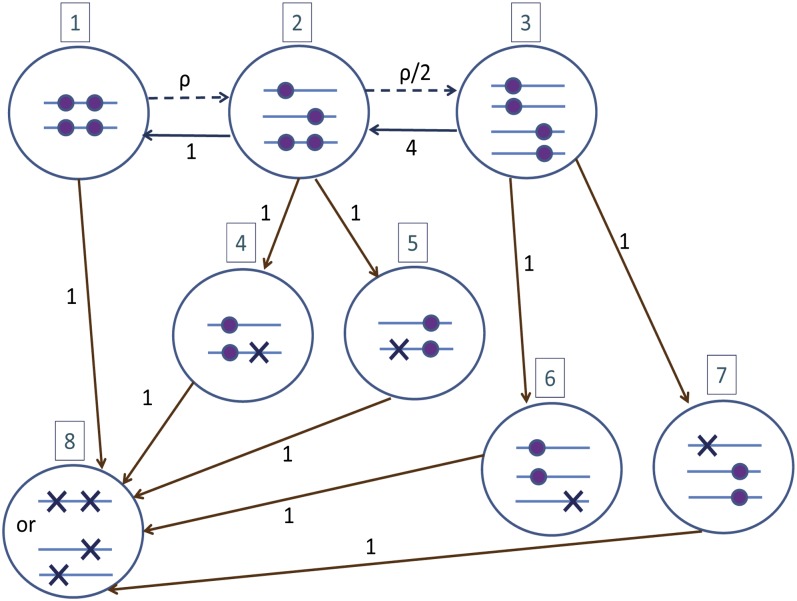
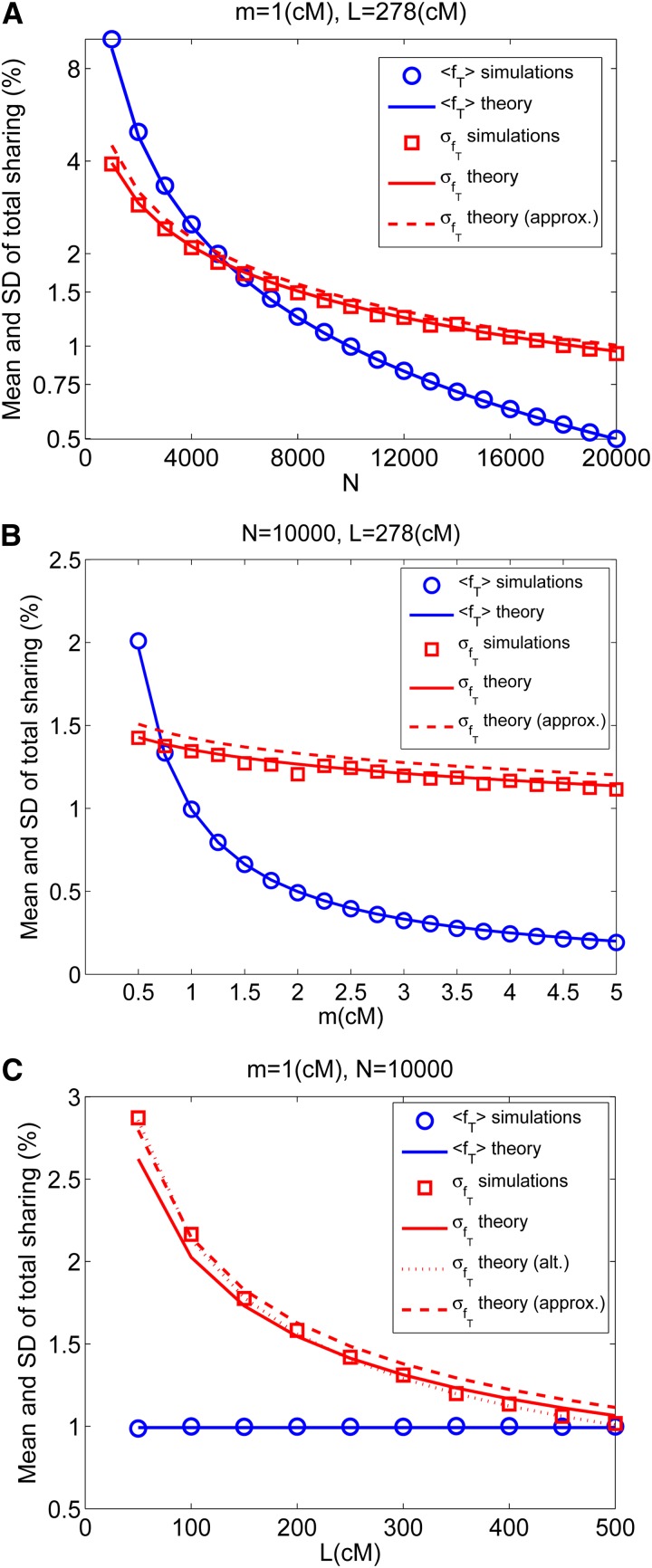
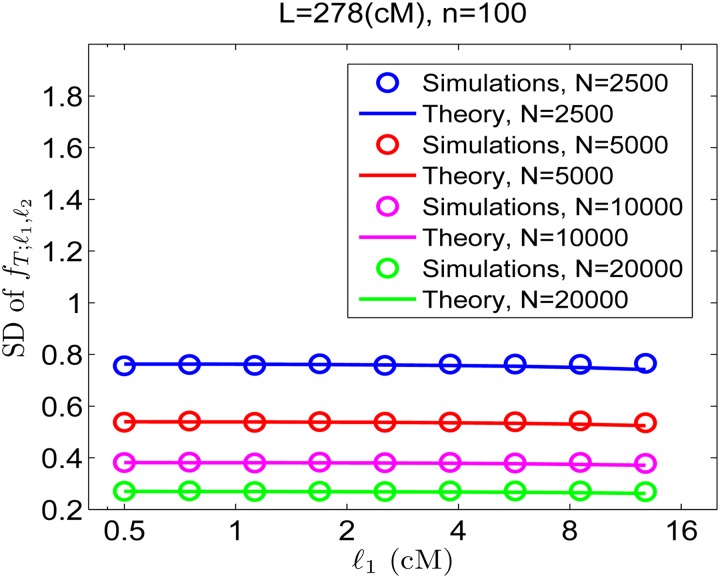
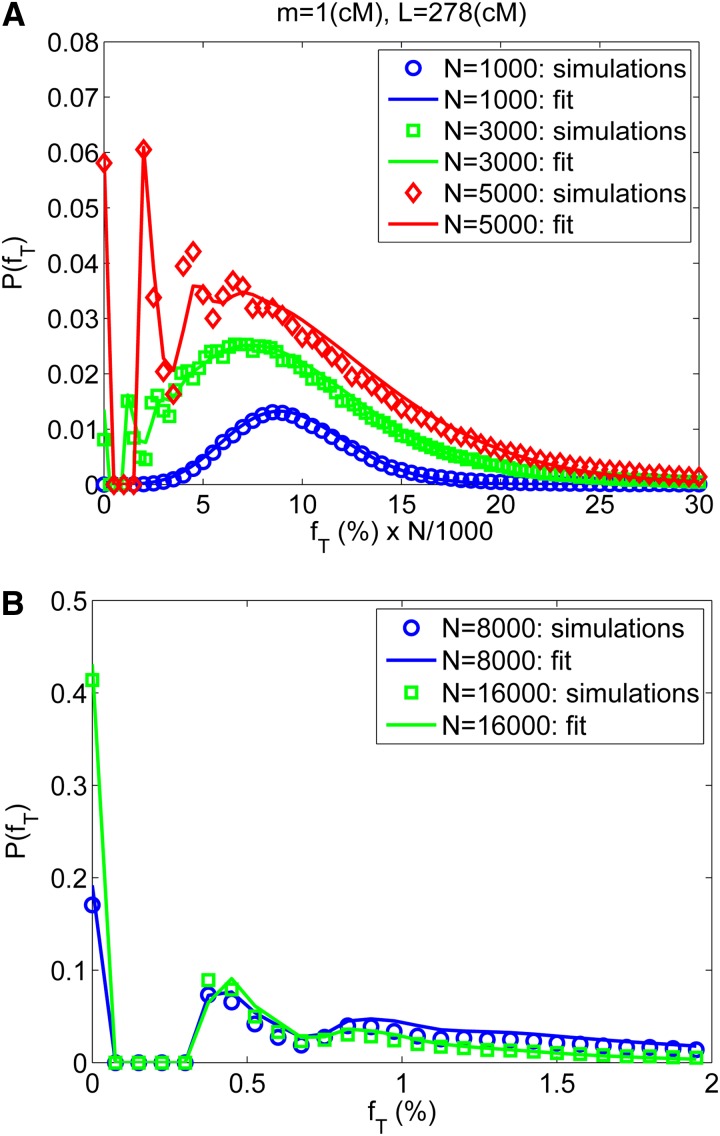
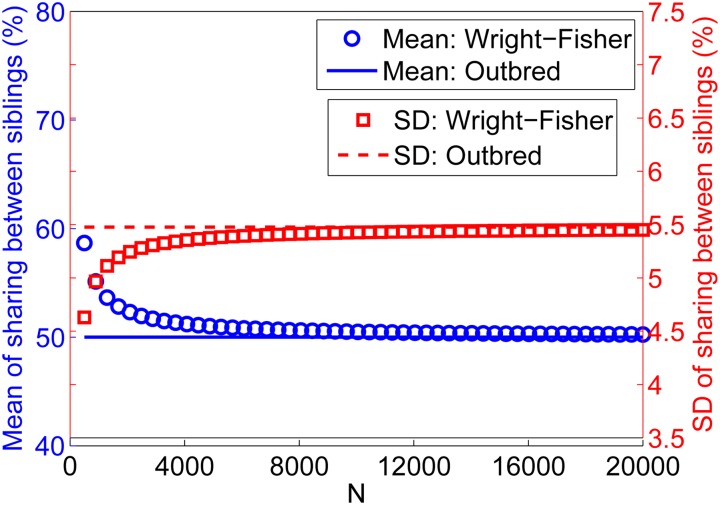
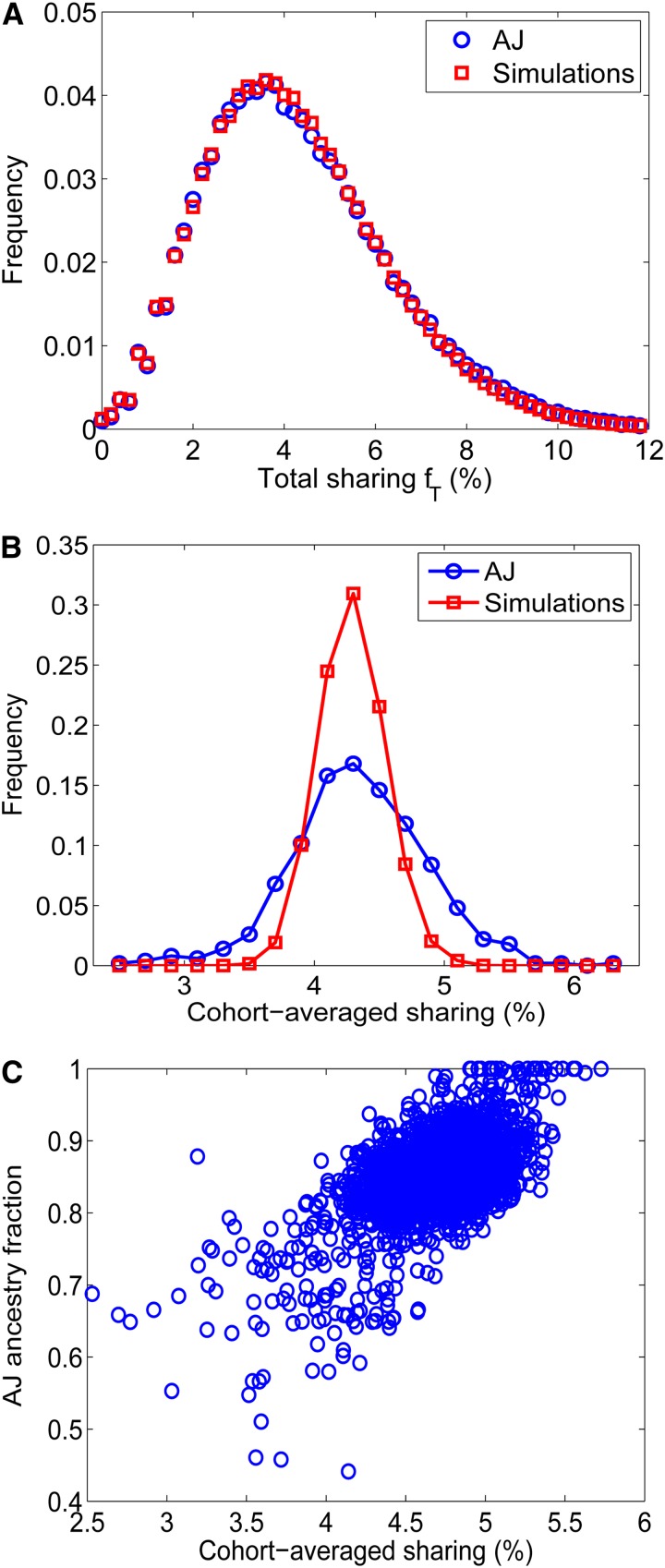
References
-
- Albrechtsen A., Korneliussen T. S., Moltke I., van Overseem Hansen T., Nielsen F. C., et al. , 2009. Relatedness mapping and tracts of relatedness for genome-wide data in the presence of linkage disequilibrium. Genet. Epidemiol. 33: 266–274. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
