

Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc
- PMID: 23267072
- PMCID: PMC3545757
- DOI: 10.1073/pnas.1218667110
Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc
Abstract
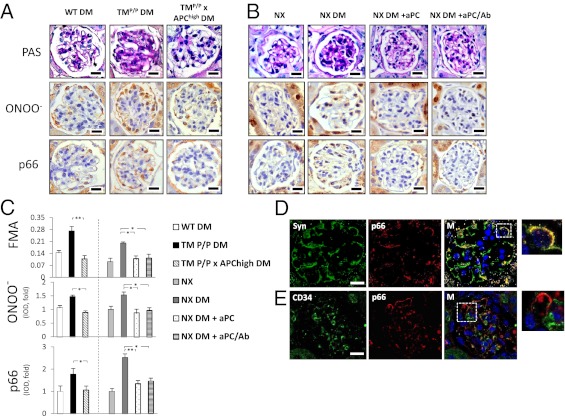
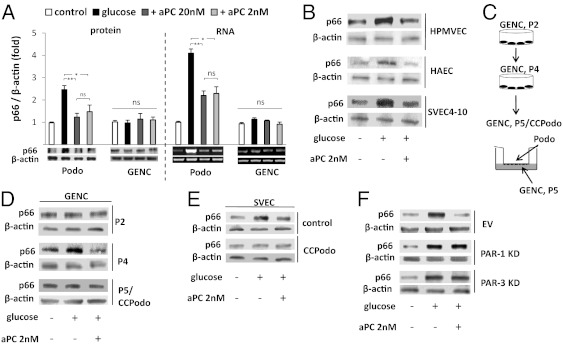
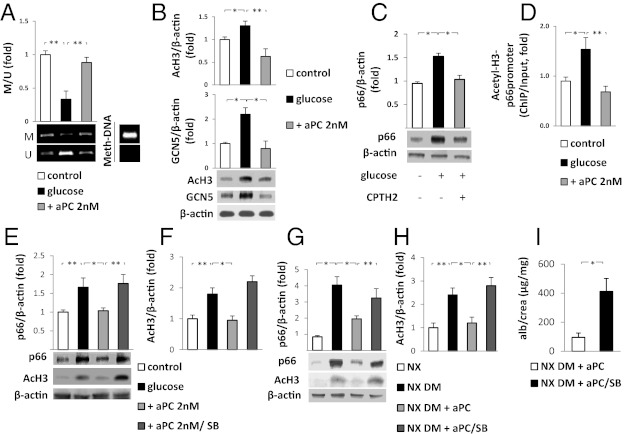
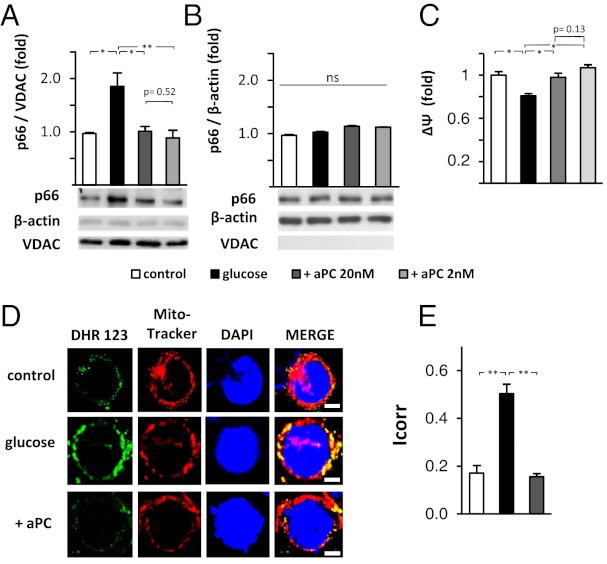
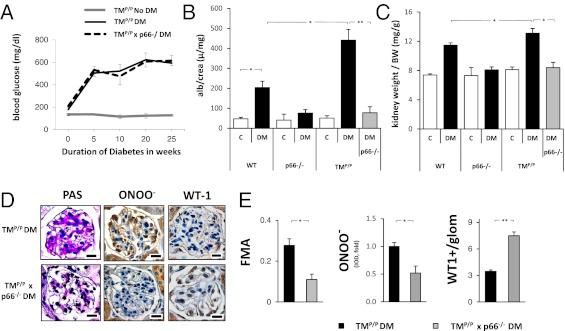
The coagulation protease activated protein C (aPC) confers cytoprotective effects in various in vitro and in vivo disease models, including diabetic nephropathy. The nephroprotective effect may be related to antioxidant effects of aPC. However, the mechanism through which aPC may convey these antioxidant effects and the functional relevance of these properties remain unknown. Here, we show that endogenous and exogenous aPC prevents glomerular accumulation of oxidative stress markers and of the redox-regulating protein p66(Shc) in experimental diabetic nephropathy. These effects were predominately observed in podocytes. In vitro, aPC inhibited glucose-induced expression of p66(Shc) mRNA and protein in podocytes (via PAR-1 and PAR-3) and various endothelial cell lines, but not in glomerular endothelial cells. Treatment with aPC reversed glucose-induced hypomethylation and hyperacetylation of the p66(Shc) promoter in podocytes. The hyperacetylating agent sodium butyrate abolished the suppressive effect of aPC on p66(Shc) expression both in vitro and in vivo. Moreover, sodium butyrate abolished the beneficial effects of aPC in experimental diabetic nephropathy. Inhibition of p66(Shc) expression and mitochondrial translocation by aPC normalized mitochondrial ROS production and the mitochondrial membrane potential in glucose-treated podocytes. Genetic ablation of p66(Shc) compensated for the loss of protein C activation in vivo, normalizing markers of diabetic nephropathy and oxidative stress. These studies identify a unique mechanism underlying the cytoprotective effect of aPC. Activated PC epigenetically controls expression of the redox-regulating protein p66(Shc), thus linking the extracellular protease aPC to mitochondrial function in diabetic nephropathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296(5574):1880–1882. - PubMed
-
- Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276(14):11199–11203. - PubMed
-
- Yamaji K, et al. Activated protein C, a natural anticoagulant protein, has antioxidant properties and inhibits lipid peroxidation and advanced glycation end products formation. Thromb Res. 2005;115(4):319–325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
