

doi: 10.1038/nmeth.2309.
Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry
Affiliations
- PMID: 23269374
- PMCID: PMC3943160
- DOI: 10.1038/nmeth.2309
Item in Clipboard
Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry
Nat Methods.
2013 Jan.
Abstract
Targeted mass spectrometry (MS) is becoming widely used in academia and in pharmaceutical and biotechnology industries for sensitive and quantitative detection of proteins, peptides and post-translational modifications. Here we describe the increasing importance of targeted MS technologies in clinical proteomics and the potential key roles these techniques will have in bridging biomedical discovery and clinical implementation.
Figures
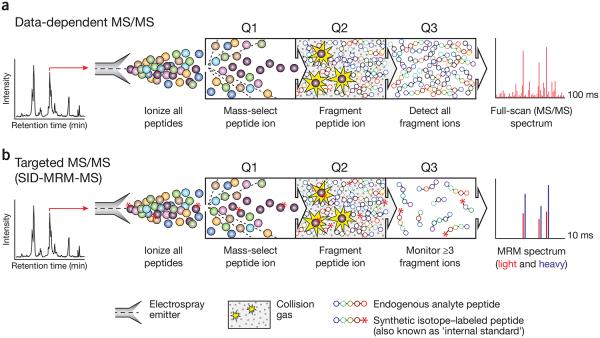
Comparison of conventional data-dependent analysis to targeted MRM-MS on a triple quadrupole mass spectrometer. (a) In a data-dependent MS experiment, digested proteins are loaded on a reversed-phase column attached to a liquid chromatography setup and eluted via electrospray to yield gas-phase ions. At any given point in the chromatographic separation many tens to hundreds of peptides are eluting nearly simultaneously. A full-scan MS spectrum is acquired and informs collection of subsequent MS/MS scans in which 4–10 ions observed in the MS spectrum are automatically selected on the basis of their signal intensity (Q1) for fragmentation by collision with inert gas (Q2). The complete array of fragment ions is detected (Q3), which constitutes the full-scan MS/MS spectrum (far right). (b) In an SID-MRM-MS analysis, proteotypic peptides uniquely representing proteins of interest are predefined together with their most informative fragment ions. Peptides are selected for fragmentation (Q1 and Q2), and fragment ions are selected for detection (Q3) based on a user-specified list of targeted precursor-fragment pairs (‘transitions’). Synthetic peptides containing stable-isotope labels can be spiked in as standards (asterisks). Comparing labeled to unlabeled peak area (far right) provides precise relative quantification of the endogenous analyte.
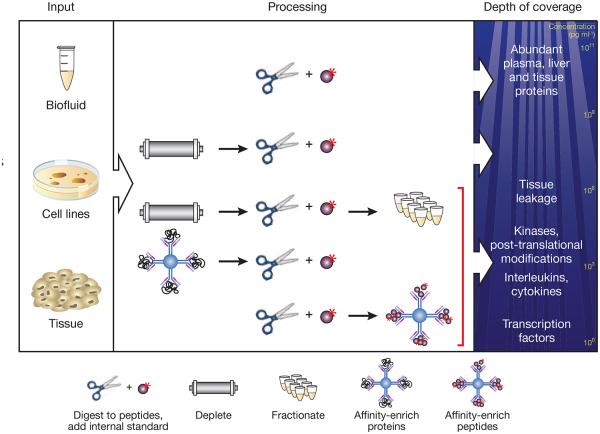
Protein and peptide enrichment strategies to increase sensitivity and specificity of analyte detection in SID-MRM-MS. After extraction from tissues, cell lines or biofluids (left), proteins are digested into peptides. To achieve the highest detection confidence and measurement precision for peptides in these complex samples, synthetic versions of each analyte peptide containing an amino acid labeled with a stable isotope (for example, 15N or 13C) are added as internal standards (top line, center). Additional strategies may be required to achieve desired detection sensitivity in complex matrices. In plasma, depletion of the 12–70 most abundant plasma proteins using immunoaffinity depletion columns can result in limits of detection of ~100 ng ml−1 (second line, center), representing ‘middling’ depths in the plasma peptide ocean (right). Biomarkers in diseases such as cancer and cardiovascular disease are commonly present in the low nanogram per milliliter range and can be robustly detected by coupling depletion to limited fractionation (third line, center), albeit at the expense of reducing throughput and introducing variability and analytical complexity. A different strategy to enhance sensitivity relies on immunoaffinity reagents to enrich target analytes. In one implementation the intact protein is captured using an anti-protein antibody before digestion (fourth line, center). Alternatively, a proteotypic peptide derived from the protein can be captured using an anti-peptide antibody to the target peptide of interest (bottom line, center).
References
-
- Lawson AM. The scope of mass spectrometry in clinical chemistry. Clin. Chem. 1975;21:803–824. - PubMed
-
- Zhu X, Desiderio DM. Peptide quantification by tandem mass spectrometry. Mass Spectrom. Rev. 1996;15:213–240. - PubMed
-
- Yost RA, Enke CG. Triple quadrupole mass spectrometry for direct mixture analysis and structure elucidation. Anal. Chem. 1979;51:1251–1264. - PubMed
-
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods. 2012;9:555–566. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
