

Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent
- PMID: 23271983
- PMCID: PMC3521656
- DOI: 10.1371/journal.pgen.1003149
Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent
Abstract
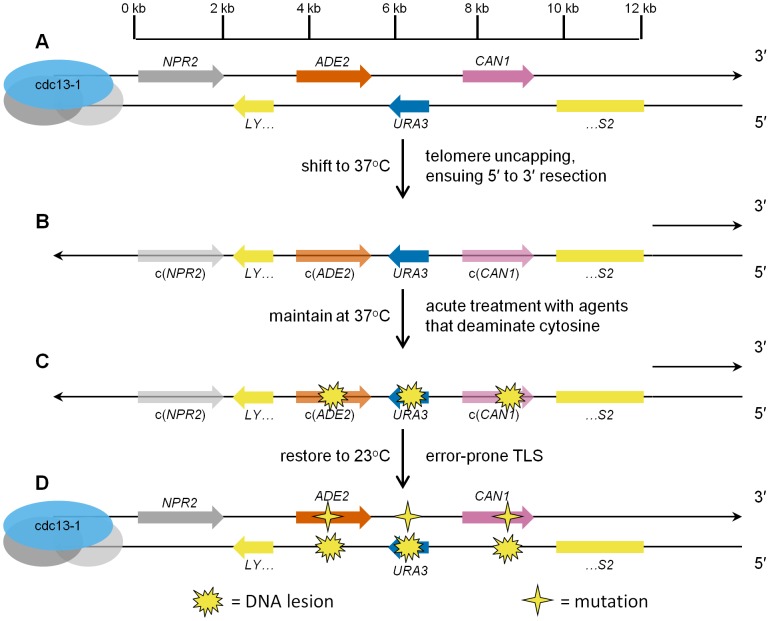
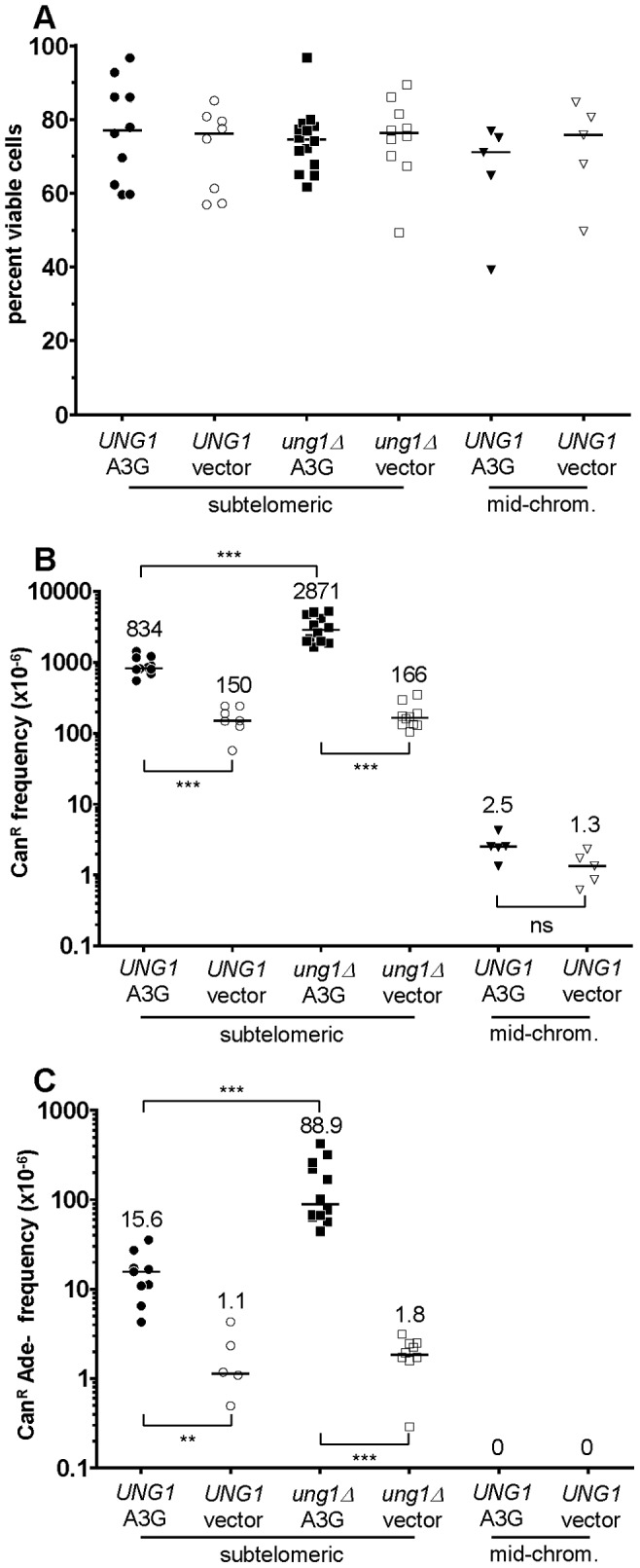
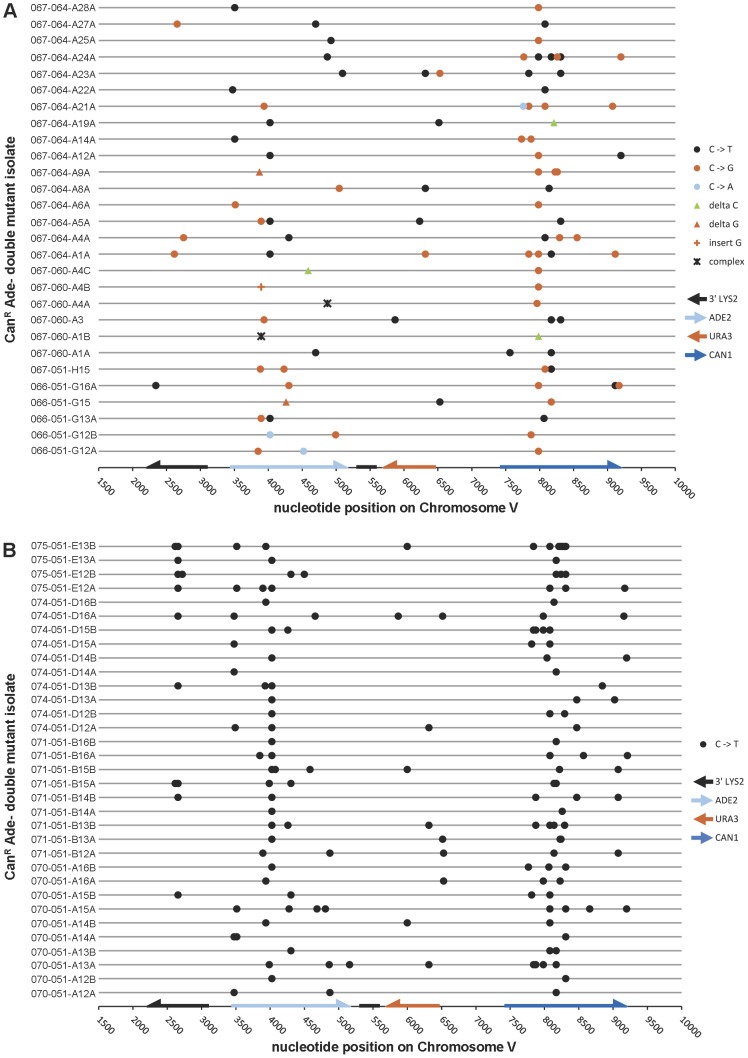
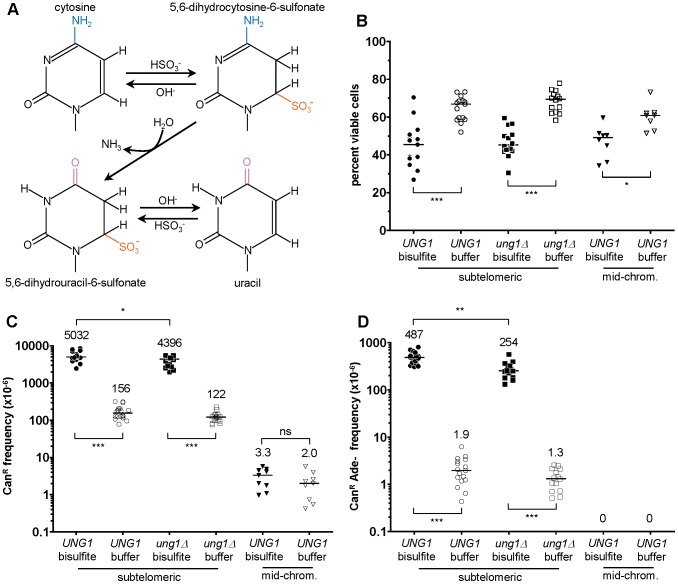
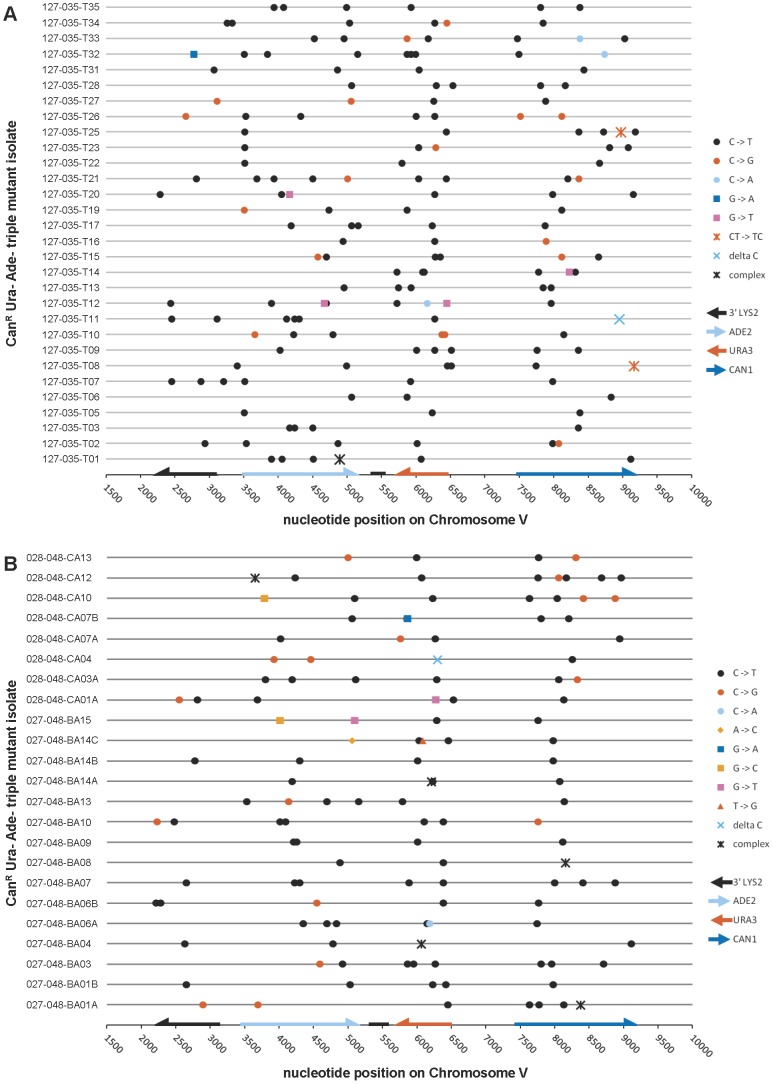
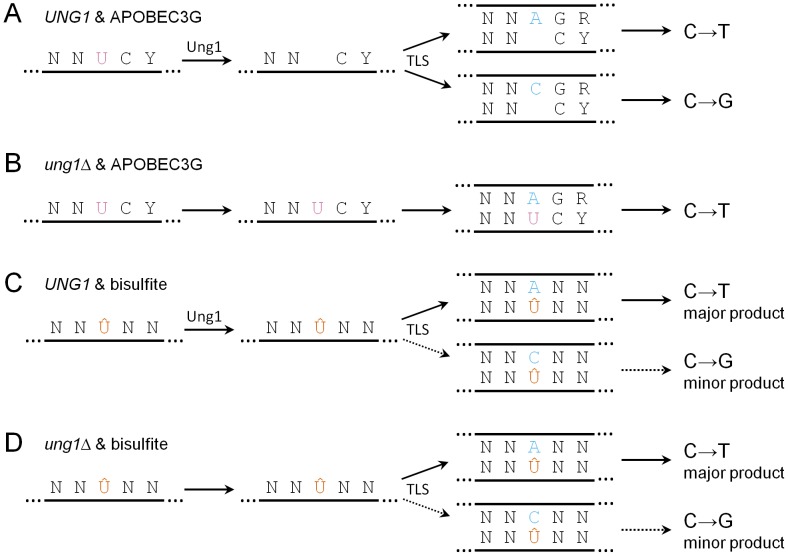
Chromosomal DNA must be in single-strand form for important transactions such as replication, transcription, and recombination to occur. The single-strand DNA (ssDNA) is more prone to damage than double-strand DNA (dsDNA), due to greater exposure of chemically reactive moieties in the nitrogenous bases. Thus, there can be agents that damage regions of ssDNA in vivo while being inert toward dsDNA. To assess the potential hazard posed by such agents, we devised an ssDNA-specific mutagenesis reporter system in budding yeast. The reporter strains bear the cdc13-1 temperature-sensitive mutation, such that shifting to 37°C results in telomere uncapping and ensuing 5' to 3' enzymatic resection. This exposes the reporter region, containing three closely-spaced reporter genes, as a long 3' ssDNA overhang. We validated the ability of the system to detect mutagenic damage within ssDNA by expressing a modified human single-strand specific cytosine deaminase, APOBEC3G. APOBEC3G induced a high density of substitutions at cytosines in the ssDNA overhang strand, resulting in frequent, simultaneous inactivation of two reporter genes. We then examined the mutagenicity of sulfites, a class of reactive sulfur oxides to which humans are exposed frequently via respiration and food intake. Sulfites, at a concentration similar to that found in some foods, induced a high density of mutations, almost always as substitutions at cytosines in the ssDNA overhang strand, resulting in simultaneous inactivation of at least two reporter genes. Furthermore, sulfites formed a long-lived adducted 2'-deoxyuracil intermediate in DNA that was resistant to excision by uracil-DNA N-glycosylase. This intermediate was bypassed by error-prone translesion DNA synthesis, frequently involving Pol ζ, during repair synthesis. Our results suggest that sulfite-induced lesions in DNA can be particularly deleterious, since cells might not possess the means to repair or bypass such lesions accurately.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
