

Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia
- PMID: 23273567
- PMCID: PMC3542462
- DOI: 10.1016/j.ajhg.2012.11.011
Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia
Abstract
Opsismodysplasia is a rare, autosomal-recessive skeletal dysplasia characterized by short stature, characteristic facial features, and in some cases severe renal phosphate wasting. We used linkage analysis and whole-genome sequencing of a consanguineous trio to discover that mutations in inositol polyphosphate phosphatase-like 1 (INPPL1) cause opsismodysplasia with or without renal phosphate wasting. Evaluation of 12 families with opsismodysplasia revealed that INPPL1 mutations explain ~60% of cases overall, including both of the families in our cohort with more than one affected child and 50% of the simplex cases.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
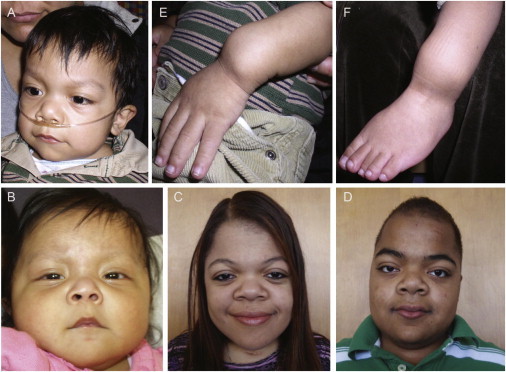
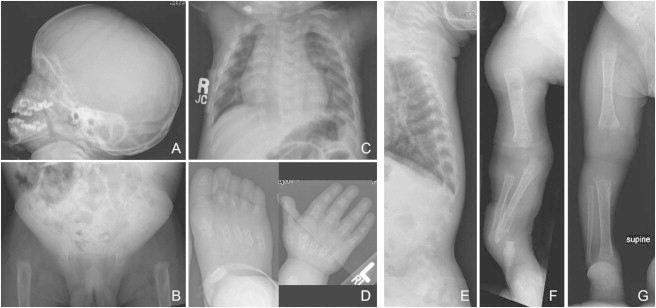
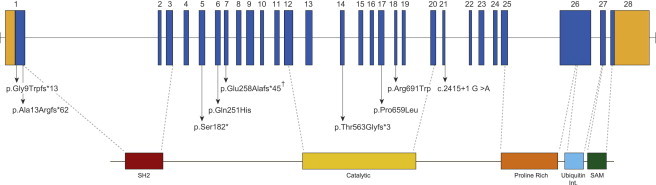
Comment in
-
Opsismodysplasia: implications of mutations in the developmental gene INPPL1.Clin Genet. 2013 Jun;83(6):527-9. doi: 10.1111/cge.12136. Epub 2013 Mar 24. Clin Genet. 2013. PMID: 23464704 No abstract available.
References
-
- Zonana J., Rimoin D.L., Lachman R.S., Cohen A.H. A unique chondrodysplasia secondary to a defect in chondroosseous transformation. Birth Defects Orig. Artic. Ser. 1977;13(3D):155–163. - PubMed
-
- Maroteaux P., Stanescu V., Stanescu R., Le Marec B., Moraine C., Lejarraga H. Opsismodysplasia: a new type of chondrodysplasia with predominant involvement of the bones of the hand and the vertebrae. Am. J. Med. Genet. 1984;19:171–182. - PubMed
-
- Cormier-Daire V., Delezoide A.L., Philip N., Marcorelles P., Casas K., Hillion Y., Faivre L., Rimoin D.L., Munnich A., Maroteaux P., Le Merrer M. Clinical, radiological, and chondro-osseous findings in opsismodysplasia: survey of a series of 12 unreported cases. J. Med. Genet. 2003;40:195–200. - PMC - PubMed
-
- Beemer F.A., Kozlowski K.S. Additional case of opsismodysplasia supporting autosomal recessive inheritance. Am. J. Med. Genet. 1994;49:344–347. - PubMed
-
- Santos H.G., Saraiva J.M. Opsismodysplasia: another case and literature review. Clin. Dysmorphol. 1995;4:222–226. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01 DE019567/DE/NIDCR NIH HHS/United States
- U54 HG006493/HG/NHGRI NIH HHS/United States
- RC2 HG005608/HG/NHGRI NIH HHS/United States
- 1RC2HG005608/HG/NHGRI NIH HHS/United States
- DE019567/DE/NIDCR NIH HHS/United States
- P01 HD022657/HD/NICHD NIH HHS/United States
- HD22657/HD/NICHD NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
- 1U54HG006493/HG/NHGRI NIH HHS/United States
- HHSN27500503415C/PHS HHS/United States
- R01 HD048895/HD/NICHD NIH HHS/United States
- HHSN267200700023C/HD/NICHD NIH HHS/United States
