

Kinetic models reveal the in vivo mechanisms of mutagenesis in microbes and man
- PMID: 23274173
- PMCID: PMC3631585
- DOI: 10.1016/j.mrrev.2012.12.003
Kinetic models reveal the in vivo mechanisms of mutagenesis in microbes and man
Abstract
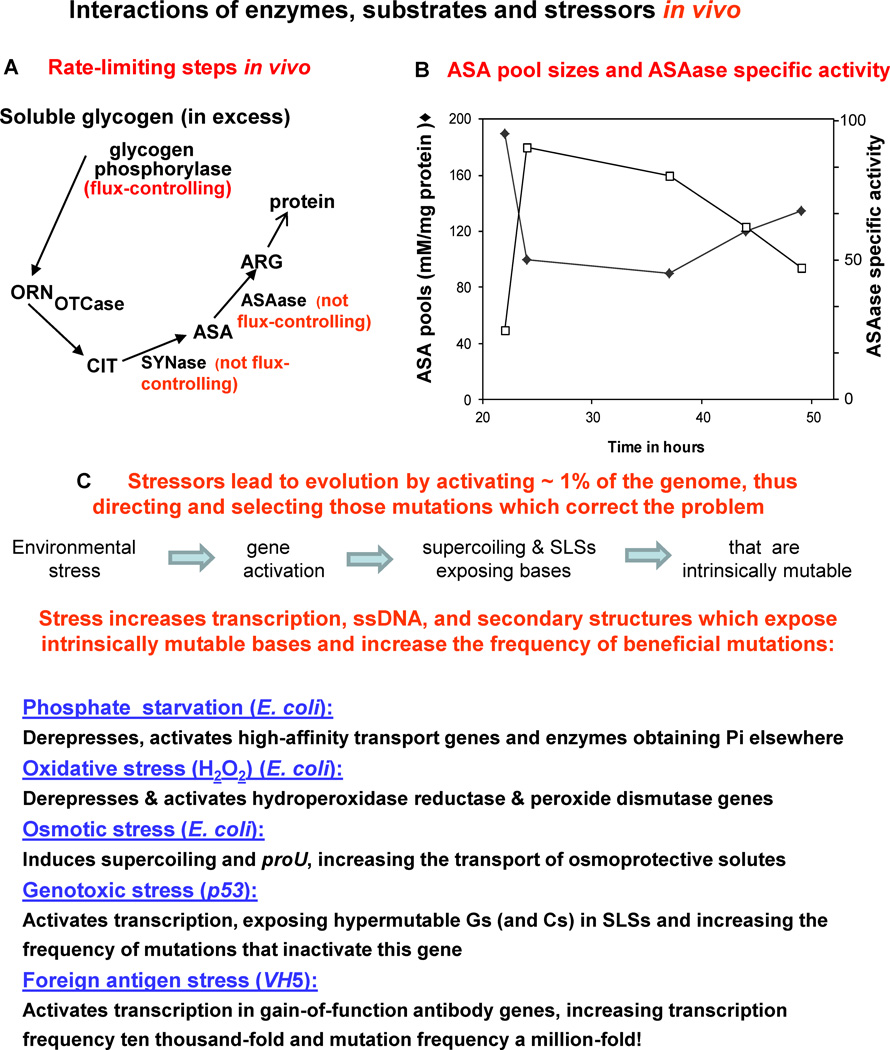
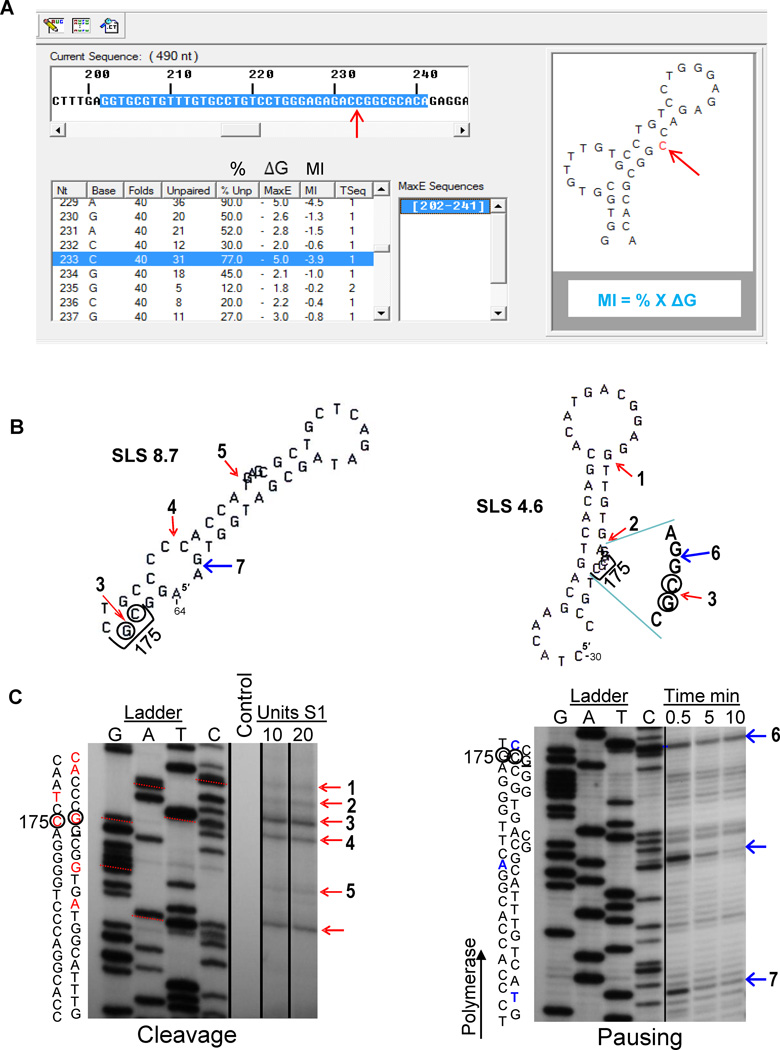
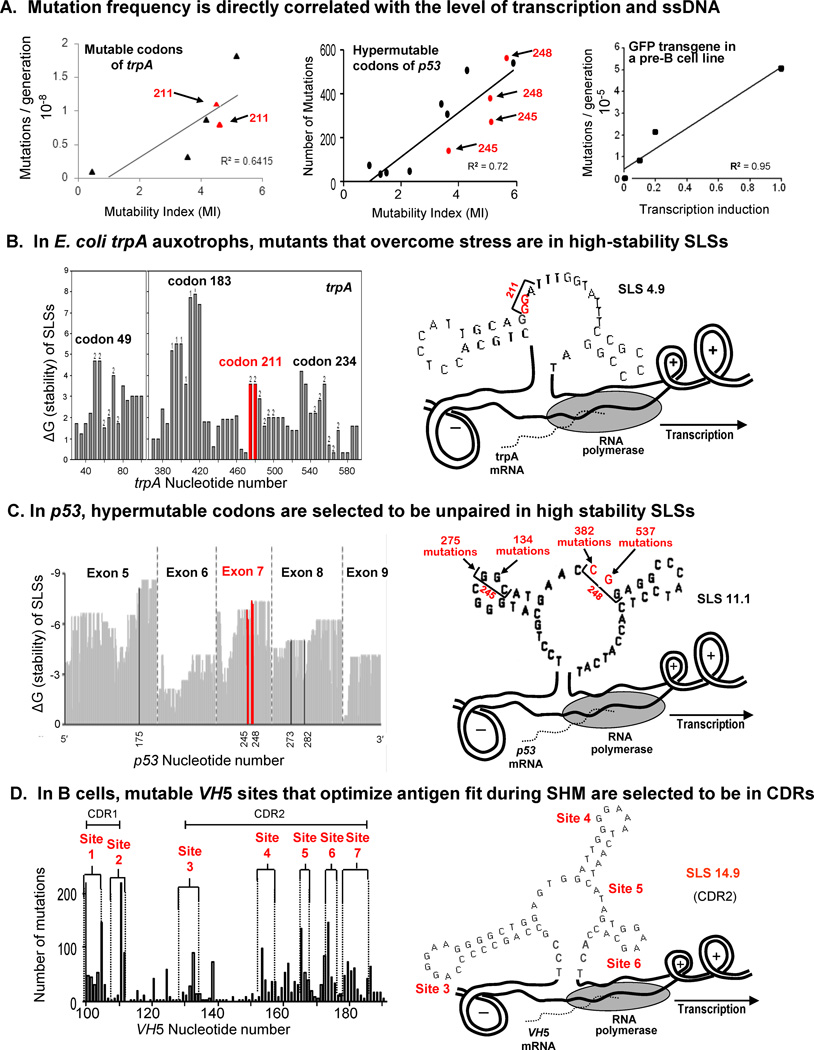
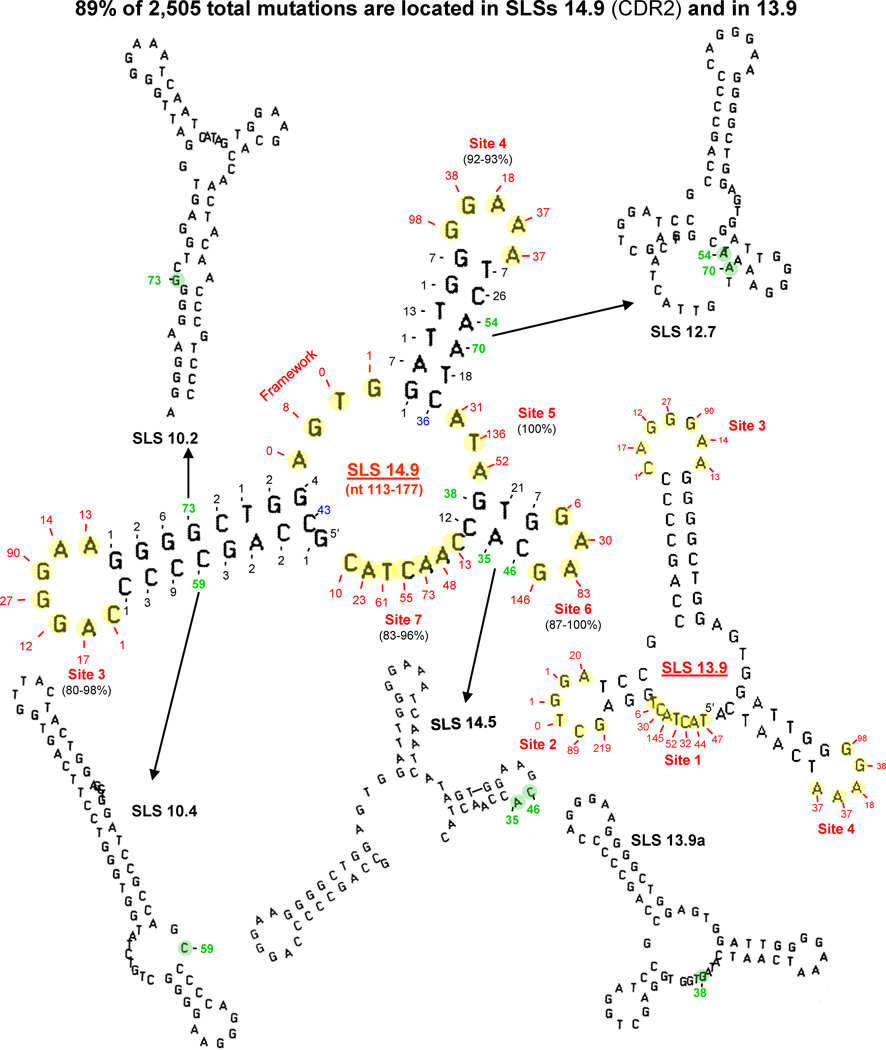
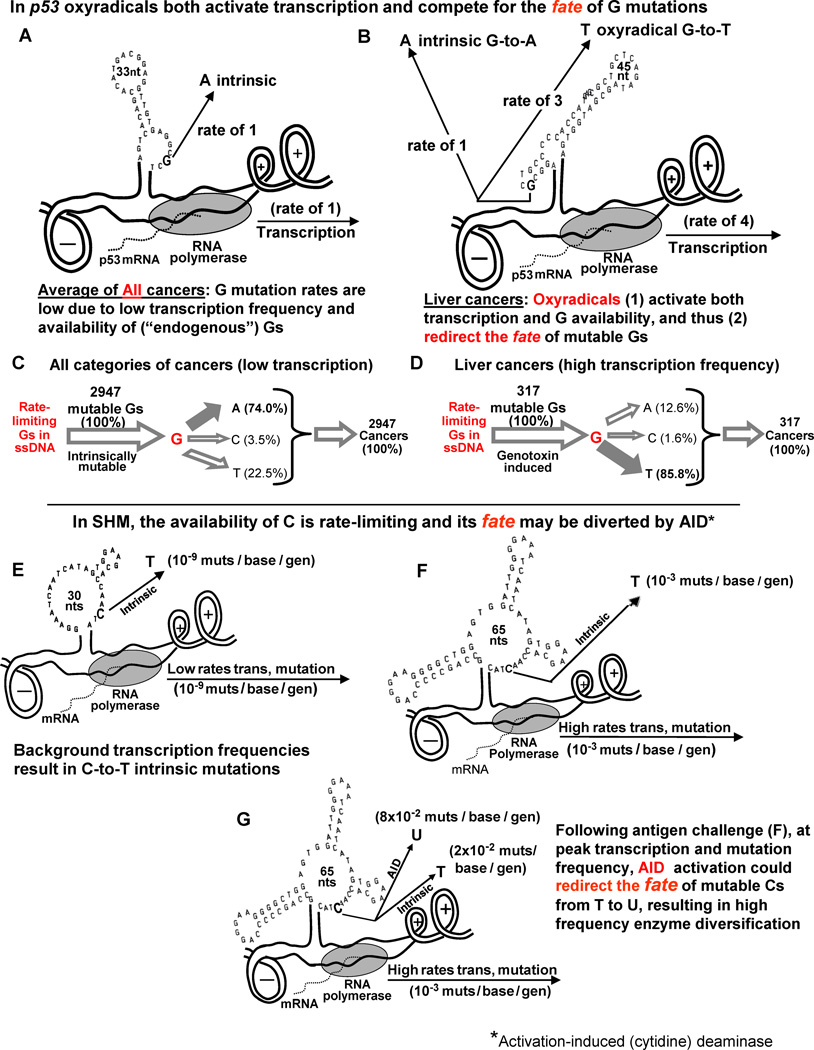
This review summarizes the evidence indicating that mutagenic mechanisms in vivo are essentially the same in all living cells. Unique metabolic reactions to a particular environmental stress apparently target specific genes for increased rates of transcription and mutation, resulting in higher mutation rates for those genes most likely to solve the problem. Kinetic models which have demonstrated predictive value are described and are shown to simulate mutagenesis in vivo in Escherichia coli, the p53 tumor suppressor gene, and somatic hypermutation. In all three models, direct correlations are seen between mutation frequencies and transcription rates. G and C nucleosides in single-stranded DNA (ssDNA) are intrinsically mutable, and G and C silent mutations in p53 and in VH framework regions provide compelling evidence for intrinsic mechanisms of mutability, since mutation outcomes are neutral and are not selected. During transcription, the availability of unpaired bases in the ssDNA of secondary structures is rate-limiting for, and determines the frequency of mutations in vivo. In vitro analyses also verify the conclusion that intrinsically mutable bases are in fact located in ssDNA loops of predicted stem-loop structures (SLSs).
Copyright © 2012 Elsevier B.V. All rights reserved.
Figures
References
-
- Kluyver AJ, Donker HJL. Die Einheit in der Biochemie. Chem d Zelle u Gew. 1926;13:134–190.
-
- Krebs H. Control of metabolic processes. Endeavour. 1957;16:125–132.
-
- Srere PA. Enzyme concentrations in tissues. Science. 1967;158:936–937. - PubMed
-
- Sauer F, Erfle JD, Binns MR. Turnover rates and intracellular pool size distribution of citrate cycle intermediates in normal, diabetic and fat-fed rats estimated by computer analysis from specific activity decay data of 14C-labeled citrate cycle acids. Eur J Biochem. 1970;17:350–363. - PubMed
-
- Hellin AC, Calmant P, Gielen J, Bours V, Merville MP. Nuclear factor – kappaB-dependent regulation of p53 gene expression induced by daunomycin genotoxic drug. Oncogene. 1998;16:1187–1195. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
