

Agalsidase benefits renal histology in young patients with Fabry disease
- PMID: 23274955
- PMCID: PMC3537211
- DOI: 10.1681/ASN.2012030316
Agalsidase benefits renal histology in young patients with Fabry disease
Abstract
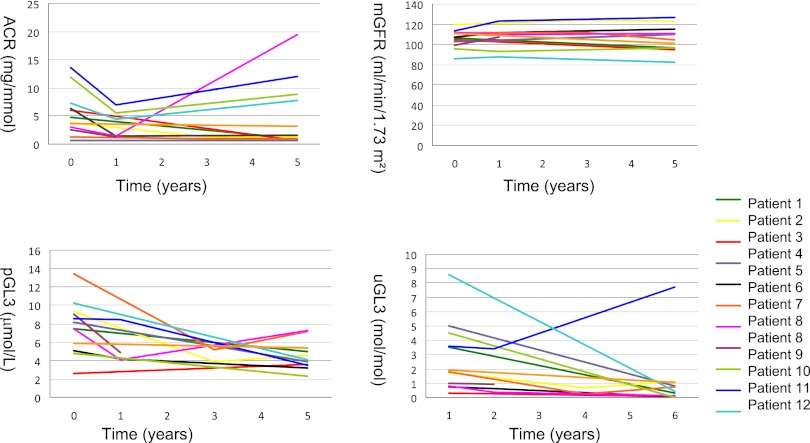
The effect of early-onset enzyme replacement therapy on renal morphologic features in Fabry disease is largely unknown. Here, we evaluated the effect of 5 years of treatment with agalsidase alfa or agalsidase beta in 12 consecutive patients age 7-33 years (median age, 16.5 years). We performed renal biopsies at baseline and after 5 years of enzyme replacement therapy; 7 patients had additional biopsies after 1 and 3 years. After a median of 65 months, biopsy findings from all patients showed total clearance of glomerular endothelial and mesangial cell inclusions, and findings from 2 patients showed complete clearance of inclusions from epithelial cells of the distal tubule. The 4 patients who received the highest dose of agalsidase exhibited substantial clearance of podocyte inclusions, and the youngest patient had nearly complete clearance of these inclusions. Linear regression analysis showed a highly significant correlation between podocyte globotriaocylceramide clearance and cumulative agalsidase dose (r=0.804; P=0.002). Microalbuminuria normalized in five patients. In summary, long-term enzyme replacement therapy in young patients can result in complete globotriaocylceramide clearance of mesangial and glomerular endothelial cells across all dosage regimens, and clearance of podocyte inclusions is dose-dependent.
Figures



Comment in
-
Fabry disease: Enzyme replacement in Fabry disease.Nat Rev Nephrol. 2013 Mar;9(3):125. doi: 10.1038/nrneph.2013.5. Epub 2013 Jan 15. Nat Rev Nephrol. 2013. PMID: 23321565 No abstract available.
References
-
- Gubler MC, Lenoir G, Grünfeld JP, Ulmann A, Droz D, Habib R: Early renal changes in hemizygous and heterozygous patients with Fabry’s disease. Kidney Int 13: 223–235, 1978 - PubMed
-
- Alroy J, Sabnis S, Kopp JB: Renal pathology in Fabry disease. J Am Soc Nephrol 13[Suppl 2] S134–S138, 2002 - PubMed
-
- Tøndel C, Bostad L, Hirth A, Svarstad E: Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis 51: 767–776, 2008 - PubMed
-
- Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, Gal A, Beck M: The early clinical phenotype of Fabry disease: A study on 35 European children and adolescents. Eur J Pediatr 162: 767–772, 2003 - PubMed
-
- Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin Iii HA, Kopp JB: Natural history of Fabry renal disease: Influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 81: 122–138, 2002 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
