

Disease-causing missense mutations in human DNA helicase disorders
- PMID: 23276657
- PMCID: PMC3640642
- DOI: 10.1016/j.mrrev.2012.12.004
Disease-causing missense mutations in human DNA helicase disorders
Abstract
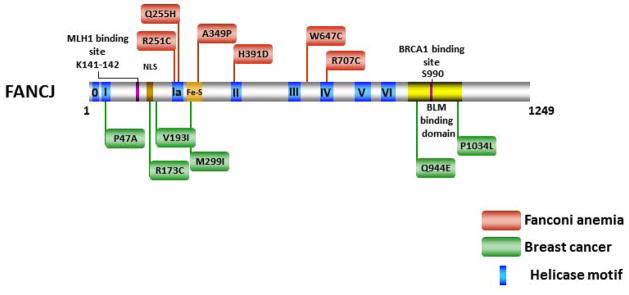
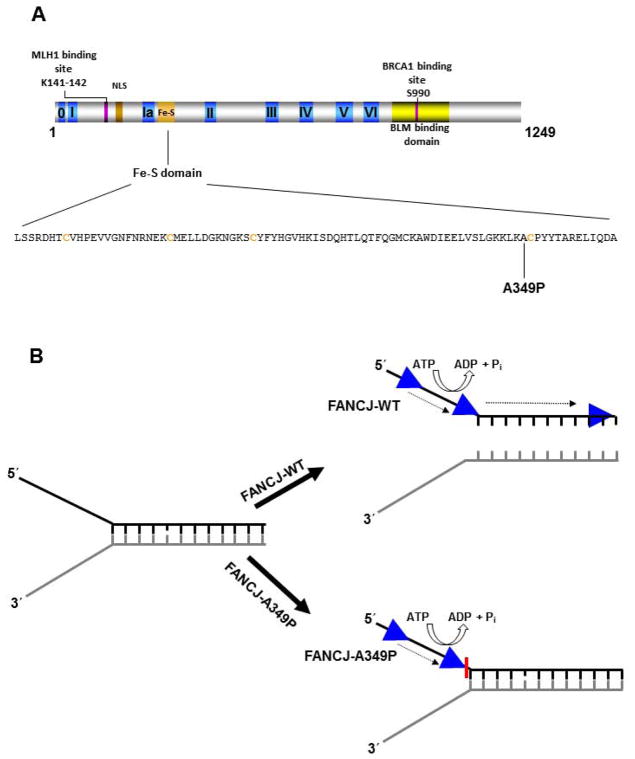
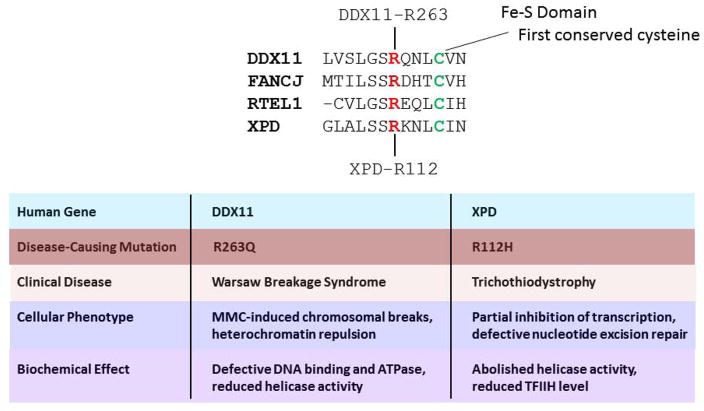
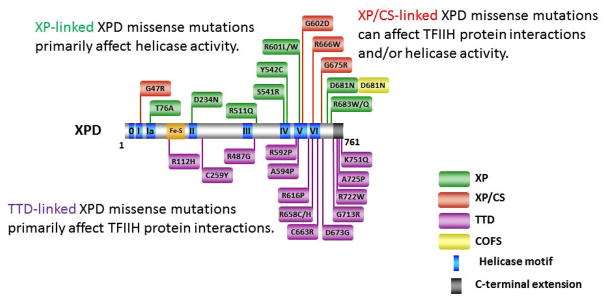
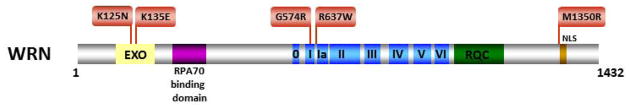
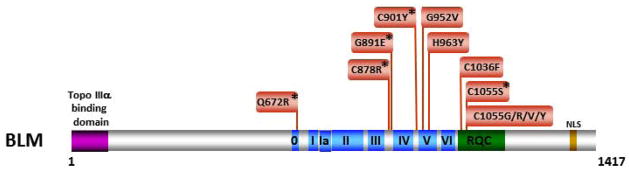
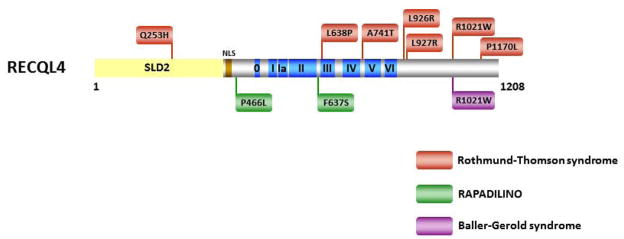
Helicases have important roles in nucleic acid metabolism, and their prominence is marked by the discovery of genetic disorders arising from disease-causing mutations. Missense mutations can yield unique insight to molecular functions and basis for disease pathology. XPB or XPD missense mutations lead to Xeroderma pigmentosum, Cockayne's syndrome, Trichothiodystrophy, or COFS syndrome, suggesting that DNA repair and transcription defects are responsible for clinical heterogeneity. Complex phenotypes are also observed for RECQL4 helicase mutations responsible for Rothmund-Thomson syndrome, Baller-Gerold syndrome, or RAPADILINO. Bloom's syndrome causing missense mutations are found in the conserved helicase and RecQ C-terminal domain of BLM that interfere with helicase function. Although rare, patient-derived missense mutations in the exonuclease or helicase domain of Werner syndrome protein exist. Characterization of WRN separation-of-function mutants may provide insight to catalytic requirements for suppression of phenotypes associated with the premature aging disorder. Characterized FANCJ missense mutations associated with breast cancer or Fanconi anemia interfere with FANCJ helicase activity required for DNA repair and the replication stress response. For example, a FA patient-derived mutation in the FANCJ Iron-Sulfur domain was shown to uncouple its ATPase and translocase activity from DNA unwinding. Mutations in DDX11 (ChlR1) are responsible for Warsaw Breakage syndrome, a recently discovered autosomal recessive cohesinopathy. Ongoing and future studies will address clinically relevant helicase mutations and polymorphisms, including those that interfere with key protein interactions or exert dominant negative phenotypes (e.g., certain mutant alleles of Twinkle mitochondrial DNA helicase). Chemical rescue may be an approach to restore helicase activity in loss-of-function helicase disorders. Genetic and biochemical analyses of disease-causing missense mutations in human helicase disorders have led to new insights to the molecular defects underlying aberrant cellular and clinical phenotypes.
Published by Elsevier B.V.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
-
- Lohman TM, Tomko EJ, Wu CG. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat Rev Mol Cell Biol. 2008;9:391–401. - PubMed
-
- Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. - PubMed
-
- Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu Rev Biochem. 1996;65:169–214. - PubMed
-
- Patel SS, Donmez I. Mechanisms of helicases. J Biol Chem. 2006;281:18625–18628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
