

Identification of a hidden strain switch provides clues to an ancient structural mechanism in protein kinases
- PMID: 23277537
- PMCID: PMC3549070
- DOI: 10.1073/pnas.1207104110
Identification of a hidden strain switch provides clues to an ancient structural mechanism in protein kinases
Abstract
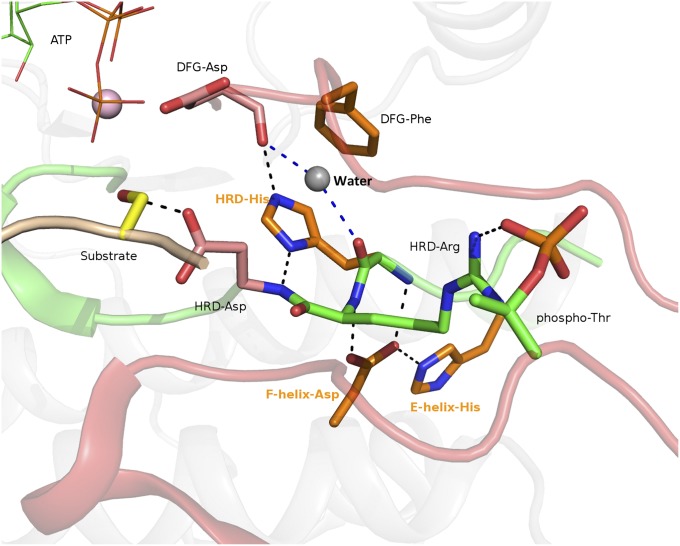
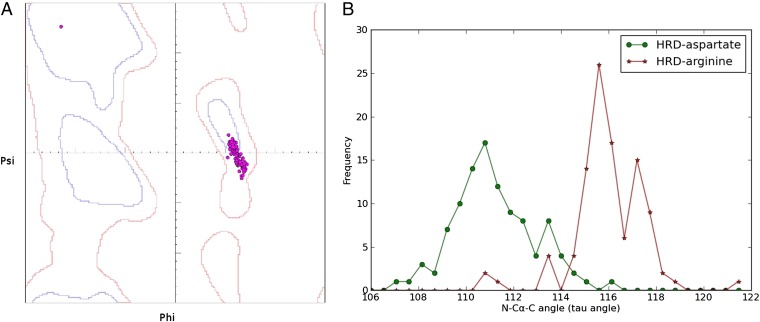
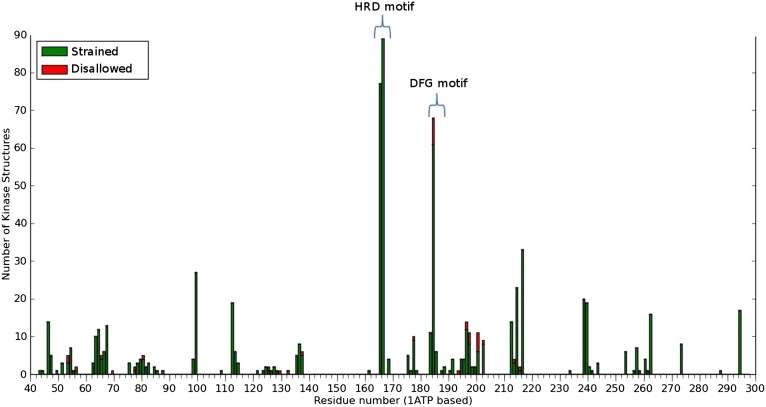
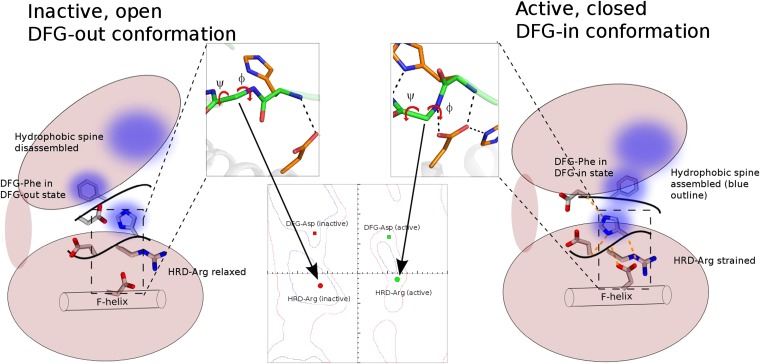
The protein kinase catalytic domain contains several conserved residues of unknown functions. Here, using a combination of computational and experimental approaches, we show that the function of some of these residues is to maintain the backbone geometry of the active site in a strained conformation. Specifically, we find that the backbone geometry of the catalytically important HRD motif deviates from ideality in high-resolution structures and the strained geometry results in favorable hydrogen bonds with conserved noncatalytic residues in the active site. In particular, a conserved aspartate in the F-helix hydrogen bonds to the strained HRD backbone in diverse eukaryotic and eukaryotic-like protein kinase crystal structures. Mutations that alter this hydrogen-bonding interaction impair catalytic activity in Aurora kinase. Although the backbone strain is present in most active conformations, several inactive conformations lack the strain because of a peptide flip in the HRD backbone. The peptide flip is correlated with loss of hydrogen bonds with the F-helix aspartate as well as with other interactions associated with kinase regulation. Within protein kinases that are regulated by activation loop phosphorylation, the strained residue is an arginine, which coordinates with the activation loop phosphate. Based on analysis of strain across the protein kinase superfamily, we propose a model in which backbone strain co-evolved with conserved residues for allosteric control of catalytic activity. Our studies provide new clues for the design of allosteric protein kinase inhibitors.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Evolutionary variation and adaptation in a conserved protein kinase allosteric network: implications for inhibitor design.Biochim Biophys Acta. 2013 Jul;1834(7):1322-9. doi: 10.1016/j.bbapap.2013.02.040. Epub 2013 Mar 14. Biochim Biophys Acta. 2013. PMID: 23499783
-
A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation.EMBO J. 2002 Oct 15;21(20):5396-407. doi: 10.1093/emboj/cdf551. EMBO J. 2002. PMID: 12374740 Free PMC article.
-
Genome-wide and structural analyses of pseudokinases encoded in the genome of Arabidopsis thaliana provide functional insights.Proteins. 2020 Dec;88(12):1620-1638. doi: 10.1002/prot.25981. Epub 2020 Aug 20. Proteins. 2020. PMID: 32667690
-
Lining the pockets of kinases and phosphatases.Curr Opin Struct Biol. 2006 Dec;16(6):693-701. doi: 10.1016/j.sbi.2006.10.006. Epub 2006 Nov 2. Curr Opin Struct Biol. 2006. PMID: 17084073 Review.
-
Evolution of the eukaryotic protein kinases as dynamic molecular switches.Philos Trans R Soc Lond B Biol Sci. 2012 Sep 19;367(1602):2517-28. doi: 10.1098/rstb.2012.0054. Philos Trans R Soc Lond B Biol Sci. 2012. PMID: 22889904 Free PMC article. Review.
Cited by
-
ProKinO: a unified resource for mining the cancer kinome.Hum Mutat. 2015 Feb;36(2):175-86. doi: 10.1002/humu.22726. Hum Mutat. 2015. PMID: 25382819 Free PMC article.
-
Hypervariability of accessible and inaccessible conformational space of proteins.Curr Res Struct Biol. 2021 Sep 16;3:229-238. doi: 10.1016/j.crstbi.2021.09.001. eCollection 2021. Curr Res Struct Biol. 2021. PMID: 34604793 Free PMC article.
-
A Dynamic Switch in Inactive p38γ Leads to an Excited State on the Pathway to an Active Kinase.Biochemistry. 2019 Dec 24;58(51):5160-5172. doi: 10.1021/acs.biochem.9b00932. Epub 2019 Dec 13. Biochemistry. 2019. PMID: 31794659 Free PMC article.
-
Hydrophobic Core Variations Provide a Structural Framework for Tyrosine Kinase Evolution and Functional Specialization.PLoS Genet. 2016 Feb 29;12(2):e1005885. doi: 10.1371/journal.pgen.1005885. eCollection 2016 Feb. PLoS Genet. 2016. PMID: 26925779 Free PMC article.
-
Relief of autoinhibition by conformational switch explains enzyme activation by a catalytically dead paralog.Elife. 2016 Dec 15;5:e20198. doi: 10.7554/eLife.20198. Elife. 2016. PMID: 27977001 Free PMC article.
References
-
- Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem Rev. 2001;101(8):2209–2242. - PubMed
-
- Krebs EG. Historical perspectives on protein phosphorylation and a classification system for protein kinases. Philos Trans R Soc Lond B Biol Sci. 1983;302(1108):3–11. - PubMed
-
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995;9(8):576–596. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
