

X-linked Charcot-Marie-Tooth disease
- PMID: 23279425
- PMCID: PMC3779456
- DOI: 10.1111/j.1529-8027.2012.00424.x
X-linked Charcot-Marie-Tooth disease
Abstract
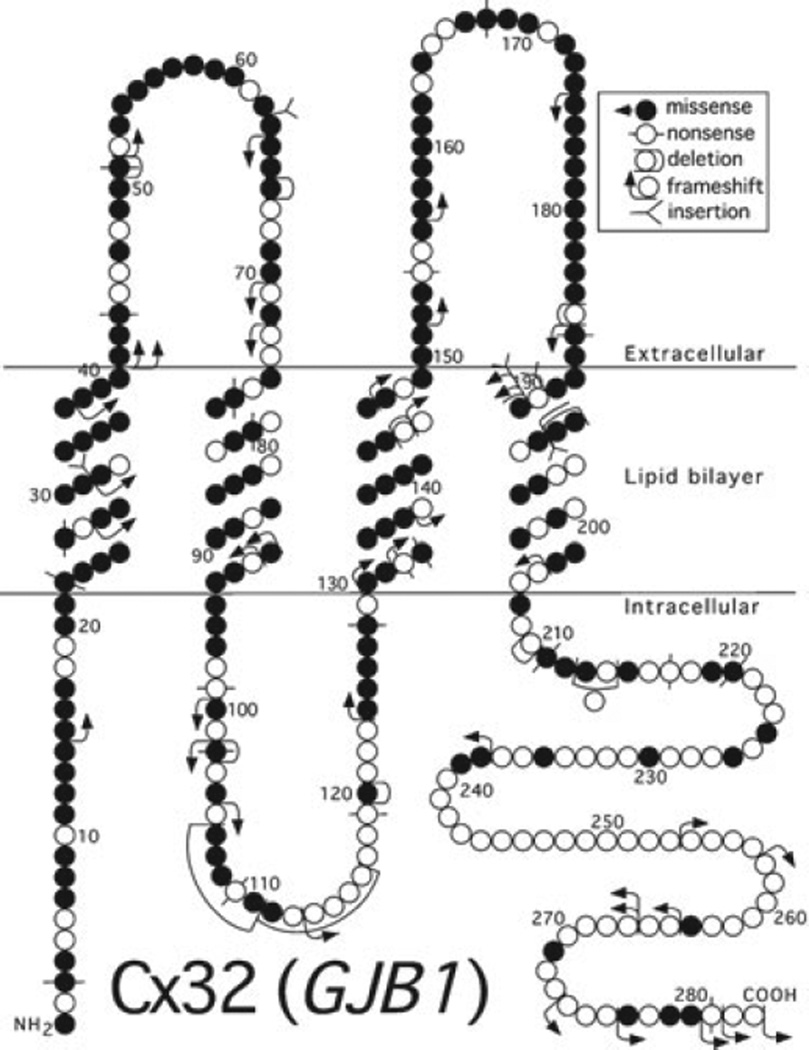
The X-linked form of Charcot-Marie-Tooth disease (CMT1X) is the second most common form of hereditary motor and sensory neuropathy. The clinical phenotype is characterized by progressive muscle atrophy and weakness, areflexia, and variable sensory abnormalities; central nervous system manifestations occur, too. Affected males have moderate to severe symptoms, whereas heterozygous females are usually less affected. Neurophysiology shows intermediate slowing of conduction and distal axonal loss. Nerve biopsies show more prominent axonal degeneration than de/remyelination. More than 400 different mutations in GJB1, the gene that encodes the gap junction (GJ) protein connexin32 (Cx32), cause CMT1X. Many Cx32 mutants fail to form functional GJs, or form GJs with abnormal biophysical properties. Schwann cells and oligodendrocytes express Cx32, and the GJs formed by Cx32 play an important role in the homeostasis of myelinated axons. Animal models of CMT1X demonstrate that loss of Cx32 in myelinating Schwann cells causes a demyelinating neuropathy. An effective therapy remains to be developed.
© 2012 Peripheral Nerve Society.
Figures
Similar articles
-
How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease?Brain Res. 2012 Dec 3;1487:198-205. doi: 10.1016/j.brainres.2012.03.068. Epub 2012 Jul 6. Brain Res. 2012. PMID: 22771394 Free PMC article. Review.
-
Molecular genetics of X-linked Charcot-Marie-Tooth disease.Neuromolecular Med. 2006;8(1-2):107-22. doi: 10.1385/nmm:8:1-2:107. Neuromolecular Med. 2006. PMID: 16775370 Review.
-
Connexins, gap junctions and peripheral neuropathy.Neurosci Lett. 2015 Jun 2;596:27-32. doi: 10.1016/j.neulet.2014.10.033. Epub 2014 Oct 24. Neurosci Lett. 2015. PMID: 25449862 Review.
-
Axonal pathology precedes demyelination in a mouse model of X-linked demyelinating/type I Charcot-Marie Tooth neuropathy.J Neuropathol Exp Neurol. 2010 Sep;69(9):945-58. doi: 10.1097/NEN.0b013e3181efa658. J Neuropathol Exp Neurol. 2010. PMID: 20720503 Free PMC article.
-
Genetic epidemiology of Charcot-Marie-Tooth disease.Acta Neurol Scand Suppl. 2012;(193):iv-22. doi: 10.1111/ane.12013. Acta Neurol Scand Suppl. 2012. PMID: 23106488
Cited by
-
Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy.Cell Rep. 2015 Aug 18;12(7):1169-83. doi: 10.1016/j.celrep.2015.07.023. Epub 2015 Aug 6. Cell Rep. 2015. PMID: 26257172 Free PMC article.
-
Connexin: a potential novel target for protecting the central nervous system?Neural Regen Res. 2015 Apr;10(4):659-66. doi: 10.4103/1673-5374.155444. Neural Regen Res. 2015. PMID: 26170830 Free PMC article. Review.
-
From BBB to PPP: Bioenergetic requirements and challenges for oligodendrocytes in health and disease.J Neurochem. 2025 Jan;169(1):e16219. doi: 10.1111/jnc.16219. Epub 2024 Sep 10. J Neurochem. 2025. PMID: 39253904 Free PMC article. Review.
-
Schwann Cells in Neuromuscular Disorders: A Spotlight on Amyotrophic Lateral Sclerosis.Cells. 2025 Jan 3;14(1):47. doi: 10.3390/cells14010047. Cells. 2025. PMID: 39791748 Free PMC article. Review.
-
The role of oligodendrocyte gap junctions in neuroinflammation.Channels (Austin). 2019 Dec;13(1):247-263. doi: 10.1080/19336950.2019.1631107. Channels (Austin). 2019. PMID: 31232168 Free PMC article. Review.
References
-
- Abel A, Bone LJ, Messing A, Scherer SS, Fischbeck KH. Studies in transgenic mice indicate a loss of connexin32 function in X-linked Charcot-Marie-Tooth disease. J Neuropathol Exp Neurol. 1999;58:702–710. - PubMed
-
- Abrams CK, Oh S, Ri Y, Bargiello TA. Mutations in connexin 32: the molecular and biophysical bases for the X-linked form of Charcot-Marie-Tooth disease. Brain Res Rev. 2000;32:203–214. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
