

New approaches for unravelling reassortment pathways
- PMID: 23279962
- PMCID: PMC3541980
- DOI: 10.1186/1471-2148-13-1
New approaches for unravelling reassortment pathways
Abstract
Background: Every year the human population encounters epidemic outbreaks of influenza, and history reveals recurring pandemics that have had devastating consequences. The current work focuses on the development of a robust algorithm for detecting influenza strains that have a composite genomic architecture. These influenza subtypes can be generated through a reassortment process, whereby a virus can inherit gene segments from two different types of influenza particles during replication. Reassortant strains are often not immediately recognised by the adaptive immune system of the hosts and hence may be the source of pandemic outbreaks. Owing to their importance in public health and their infectious ability, it is essential to identify reassortant influenza strains in order to understand the evolution of this virus and describe reassortment pathways that may be biased towards particular viral segments. Phylogenetic methods have been used traditionally to identify reassortant viruses. In many studies up to now, the assumption has been that if two phylogenetic trees differ, it is because reassortment has caused them to be different. While phylogenetic incongruence may be caused by real differences in evolutionary history, it can also be the result of phylogenetic error. Therefore, we wish to develop a method for distinguishing between topological inconsistency that is due to confounding effects and topological inconsistency that is due to reassortment.
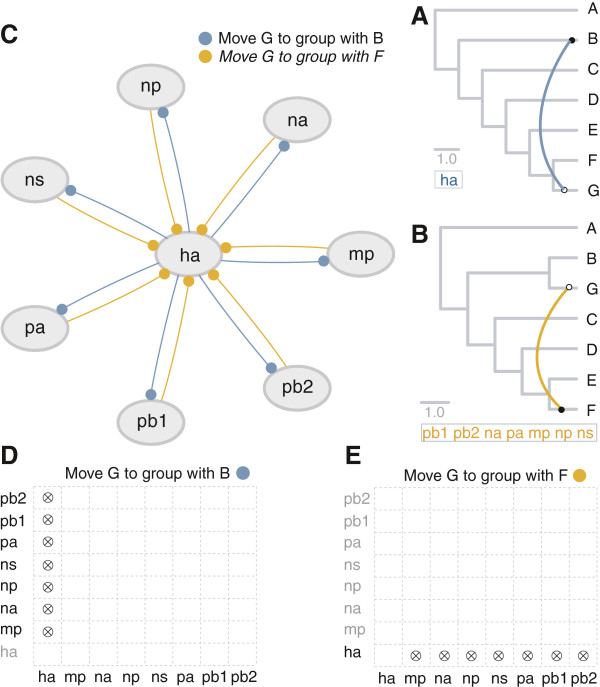
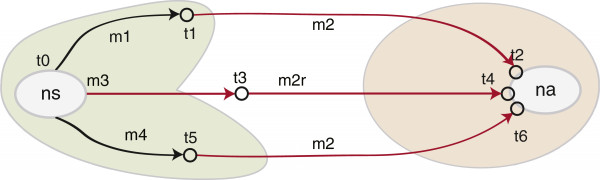
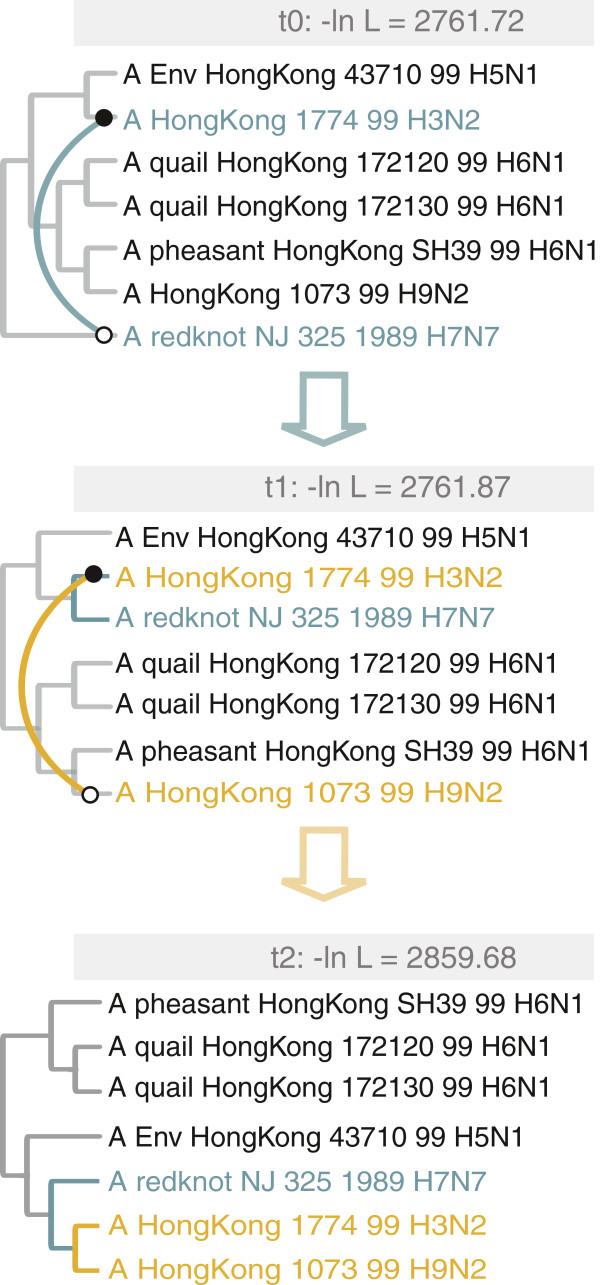
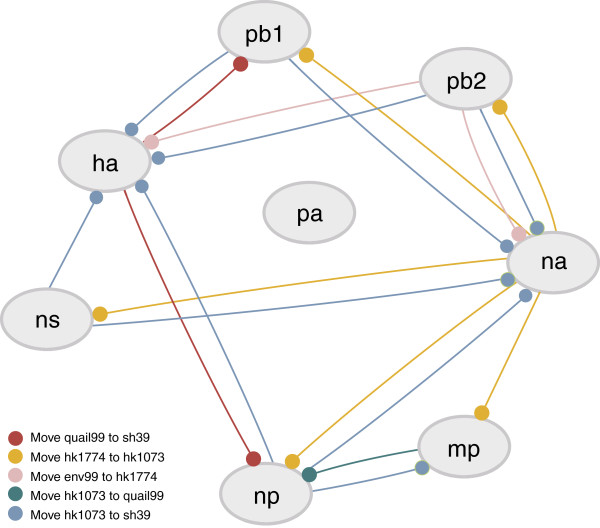
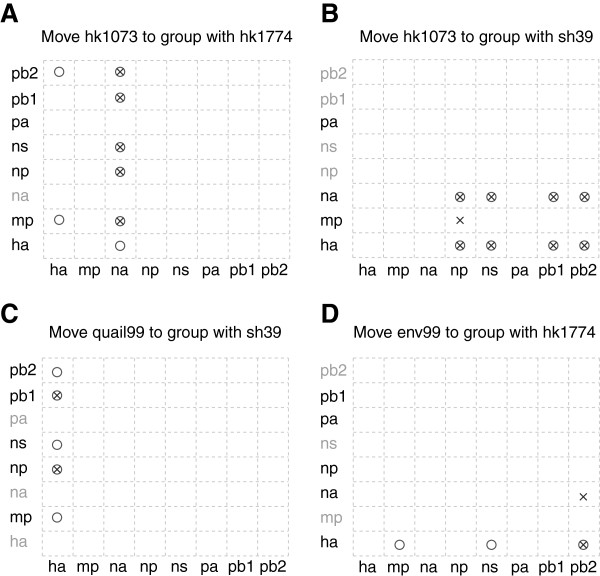
Results: The current work describes the implementation of two approaches for robustly identifying reassortment events. The algorithms rest on the idea of significance of difference between phylogenetic trees or phylogenetic tree sets, and subtree pruning and regrafting operations, which mimic the effect of reassortment on tree topologies. The first method is based on a maximum likelihood (ML) framework (MLreassort) and the second implements a Bayesian approach (Breassort) for reassortment detection. We focus on reassortment events that are found by both methods. We test both methods on a simulated dataset and on a small collection of real viral data isolated in Hong Kong in 1999.
Conclusions: The nature of segmented viral genomes present many challenges with respect to disease. The algorithms developed here can effectively identify reassortment events in small viral datasets and can be applied not only to influenza but also to other segmented viruses. Owing to computational demands of comparing tree topologies, further development in this area is necessary to allow their application to larger datasets.
Figures

Similar articles
-
A phylogenetic approach to detecting reassortments in viruses with segmented genomes.Gene. 2010 Sep 15;464(1-2):11-6. doi: 10.1016/j.gene.2010.05.002. Epub 2010 May 28. Gene. 2010. PMID: 20546849
-
Bayesian inference of reassortment networks reveals fitness benefits of reassortment in human influenza viruses.Proc Natl Acad Sci U S A. 2020 Jul 21;117(29):17104-17111. doi: 10.1073/pnas.1918304117. Epub 2020 Jul 6. Proc Natl Acad Sci U S A. 2020. PMID: 32631984 Free PMC article.
-
Detecting transmission and reassortment events for influenza A viruses with genotype profile method.Virol J. 2011 Aug 9;8:395. doi: 10.1186/1743-422X-8-395. Virol J. 2011. PMID: 21824442 Free PMC article.
-
In vivo reassortment of influenza viruses.Acta Biochim Pol. 2014;61(3):427-31. Epub 2014 Sep 3. Acta Biochim Pol. 2014. PMID: 25180223 Review.
-
Enhancement of influenza virus transmission by gene reassortment.Curr Top Microbiol Immunol. 2014;385:185-204. doi: 10.1007/82_2014_389. Curr Top Microbiol Immunol. 2014. PMID: 25048543 Review.
Cited by
-
Extensive Admixture Among Karst-Obligate Salamanders Reveals Evidence of Recent Divergence and Gene Exchange Through Aquifers.Ecol Evol. 2025 Jan 9;15(1):e70785. doi: 10.1002/ece3.70785. eCollection 2025 Jan. Ecol Evol. 2025. PMID: 39803198 Free PMC article.
-
Sexual Dimorphism in Fin Size and Shape in North American Killifish.Ecol Evol. 2025 Apr 11;15(4):e71194. doi: 10.1002/ece3.71194. eCollection 2025 Apr. Ecol Evol. 2025. PMID: 40225885 Free PMC article.
-
RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion.PLoS Pathog. 2015 Jul 9;11(7):e1004902. doi: 10.1371/journal.ppat.1004902. eCollection 2015 Jul. PLoS Pathog. 2015. PMID: 26158697 Free PMC article. Review. No abstract available.
-
TreeKnit: Inferring ancestral reassortment graphs of influenza viruses.PLoS Comput Biol. 2022 Aug 19;18(8):e1010394. doi: 10.1371/journal.pcbi.1010394. eCollection 2022 Aug. PLoS Comput Biol. 2022. PMID: 35984845 Free PMC article.
-
Chromosome-level echidna genome illuminates evolution of multiple sex chromosome system in monotremes.Gigascience. 2025 Jan 6;14:giae112. doi: 10.1093/gigascience/giae112. Gigascience. 2025. PMID: 39778707 Free PMC article.
References
-
- Laver WG, Webster RG. Studies on the origin of pandemic influenza. II. Peptide maps of the light and heavy polypeptide chains from the hemagglutinin subunits of A 2 influenza viruses isolated before and after the appearance of Hong Kong influenza. Virology. 1972;48(2):445–455. doi: 10.1016/0042-6822(72)90055-4. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
