

Mutations in KCND3 cause spinocerebellar ataxia type 22
- PMID: 23280837
- PMCID: PMC4085146
- DOI: 10.1002/ana.23701
Mutations in KCND3 cause spinocerebellar ataxia type 22
Abstract
Objective: To identify the causative gene in spinocerebellar ataxia (SCA) 22, an autosomal dominant cerebellar ataxia mapped to chromosome 1p21-q23.
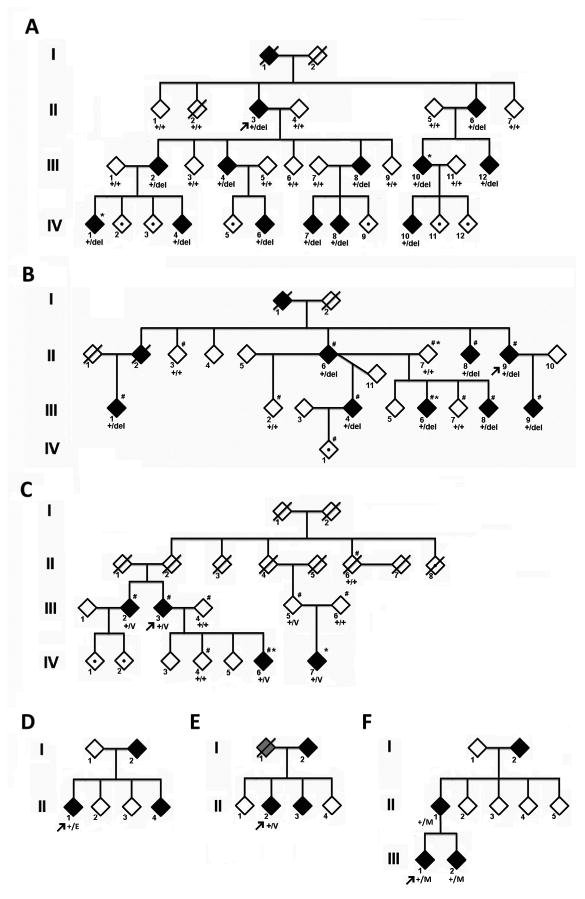
Methods: We previously characterized a large Chinese family with progressive ataxia designated SCA22, which overlaps with the locus of SCA19. The disease locus in a French family and an Ashkenazi Jewish American family was also mapped to this region. Members from all 3 families were enrolled. Whole exome sequencing was performed to identify candidate mutations, which were narrowed by linkage analysis and confirmed by Sanger sequencing and cosegregation analyses. Mutational analyses were also performed in 105 Chinese and 55 Japanese families with cerebellar ataxia. Mutant gene products were examined in a heterologous expression system to address the changes in protein localization and electrophysiological functions.
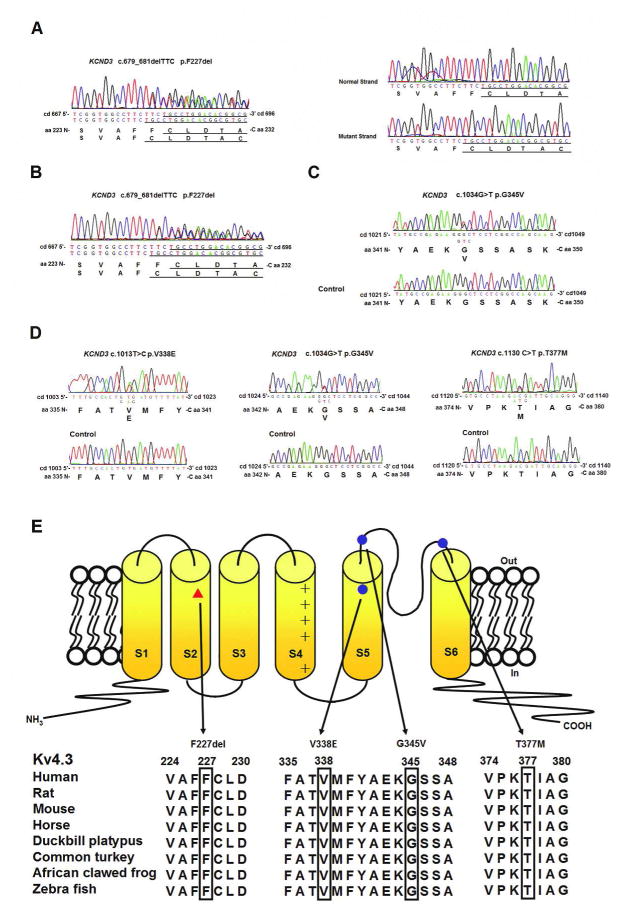
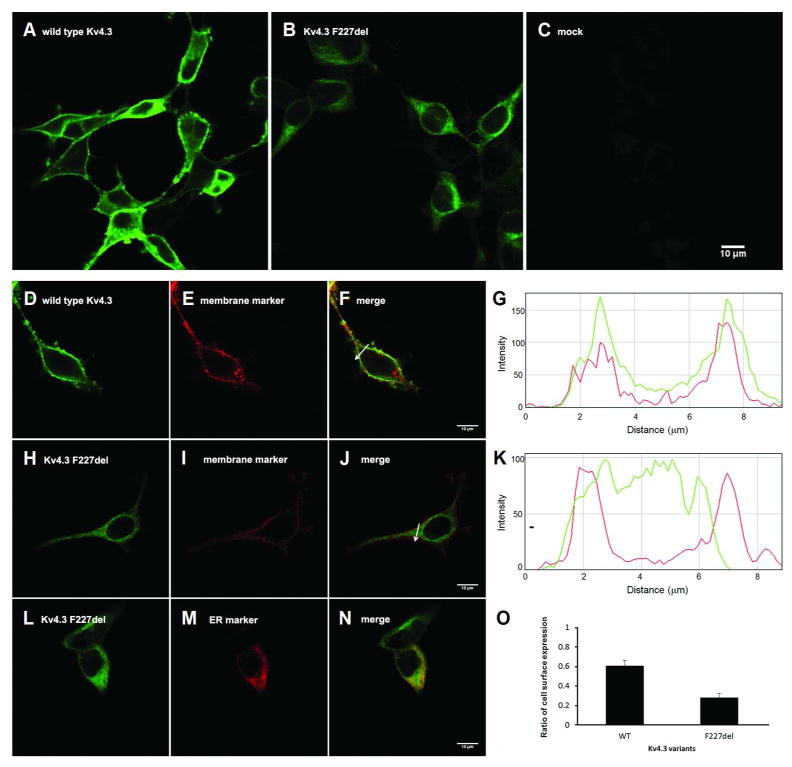
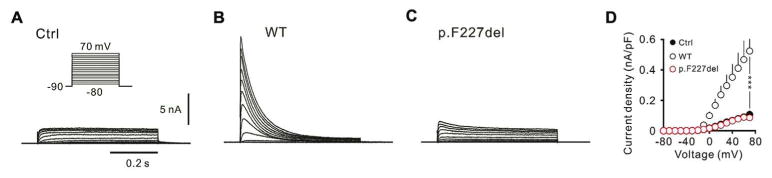
Results: We identified heterozygous mutations in the voltage-gated potassium channel Kv4.3-encoding gene KCND3: an in-frame 3-nucleotide deletion c.679_681delTTC p.F227del in both the Chinese and French pedigrees, and a missense mutation c.1034G>T p.G345V in the Ashkenazi Jewish family. Direct sequencing of KCND3 further identified 3 mutations, c.1034G>T p.G345V, c.1013T>C p.V338E, and c.1130C>T p.T377M, in 3 Japanese kindreds. Immunofluorescence analyses revealed that the mutant p.F227del Kv4.3 subunits were retained in the cytoplasm, consistent with the lack of A-type K(+) channel conductance in whole cell patch-clamp recordings.
Interpretation: Our data identify the cause of SCA19/22 in patients of diverse ethnic origins as mutations in KCND3. These findings further emphasize the important role of ion channels as key regulators of neuronal excitability in the pathogenesis of cerebellar degeneration.
Copyright © 2012 American Neurological Association.
Conflict of interest statement
Figures
Comment in
-
Genetics: Mutations in KCND3 linked to spinocerebellar ataxias.Nat Rev Neurol. 2012 Sep;8(9):472. doi: 10.1038/nrneurol.2012.165. Epub 2012 Aug 14. Nat Rev Neurol. 2012. PMID: 22890214 No abstract available.
-
Repolarization matters: mutations in the Kv4.3 potassium channel cause SCA19/22.Ann Neurol. 2012 Dec;72(6):829-31. doi: 10.1002/ana.23803. Ann Neurol. 2012. PMID: 23280833 No abstract available.
References
-
- Klockgether T. Update on degenerative ataxias. Curr Opin Neurol. 2011;24:339–345. - PubMed
-
- Soong Bw, Paulson HL. Spinocerebellar ataxias: an update. Curr Opin Neurol. 2007;20:438–446. - PubMed
-
- Soong BW, LU YC, Choo KB, Lee HY. Frequency analysis of autosomal dominant ataxias in Taiwanese patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Arch Neurol. 2001;58:1105–1109. - PubMed
-
- Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. - PubMed
-
- Chung MY, Lu YC, Cheng NC, Soong BW. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain. 2003;126:1293–1299. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
