

DNA damage drives accelerated bone aging via an NF-κB-dependent mechanism
- PMID: 23281008
- PMCID: PMC3662975
- DOI: 10.1002/jbmr.1851
DNA damage drives accelerated bone aging via an NF-κB-dependent mechanism
Abstract
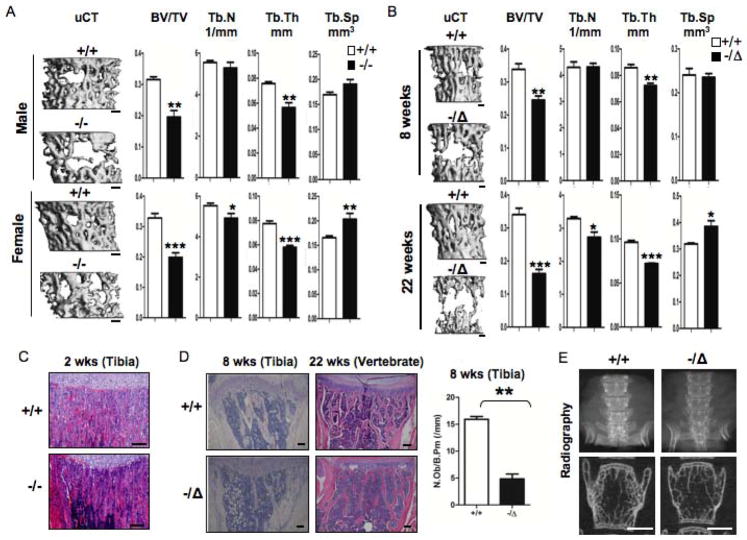
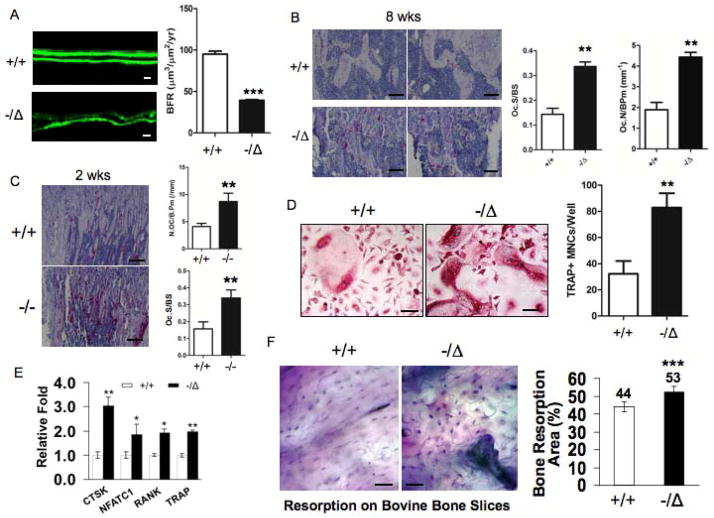
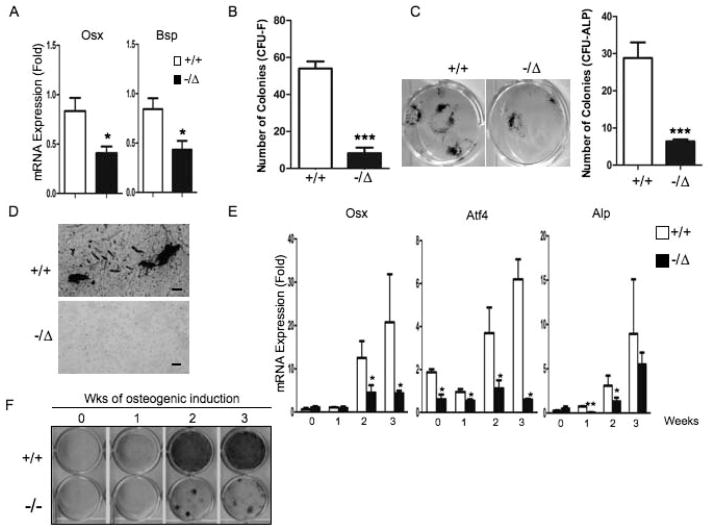
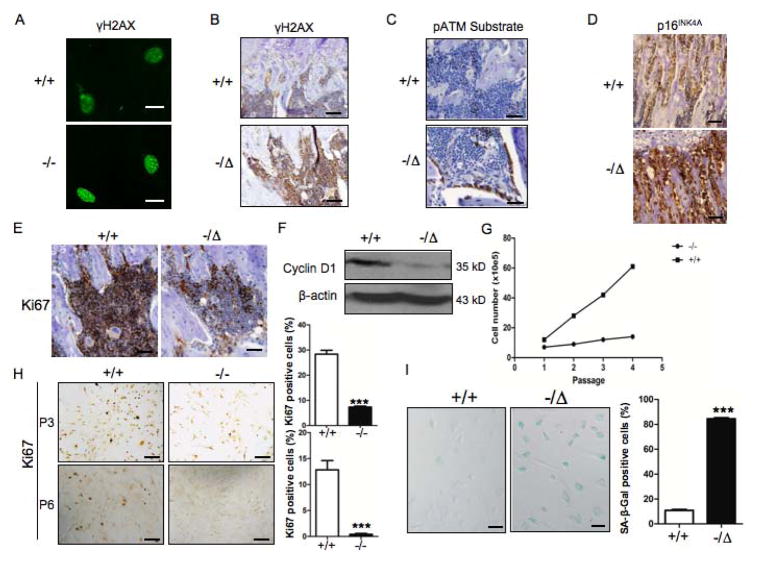
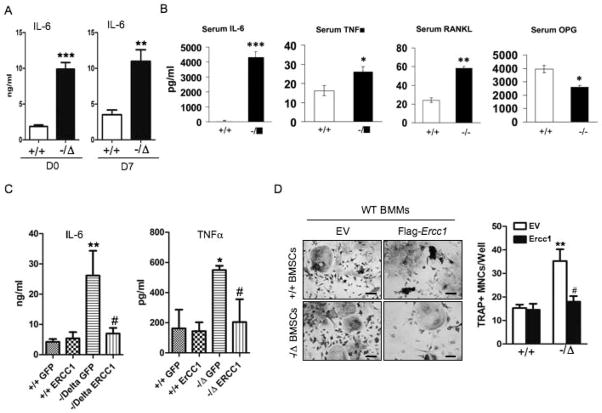
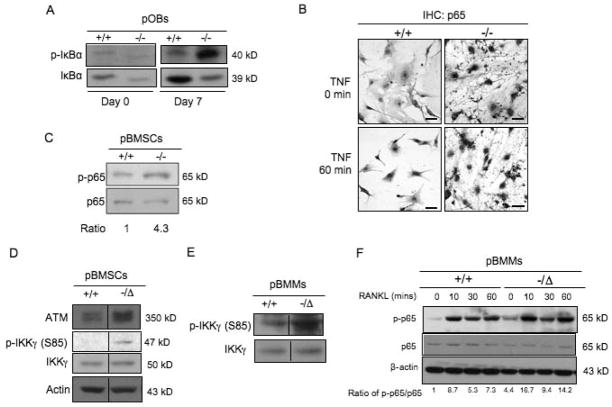
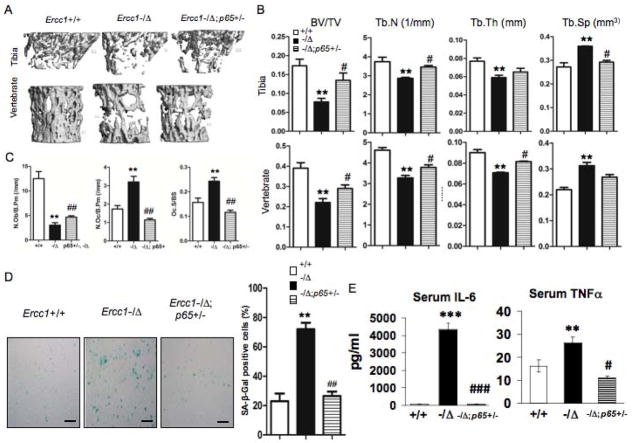
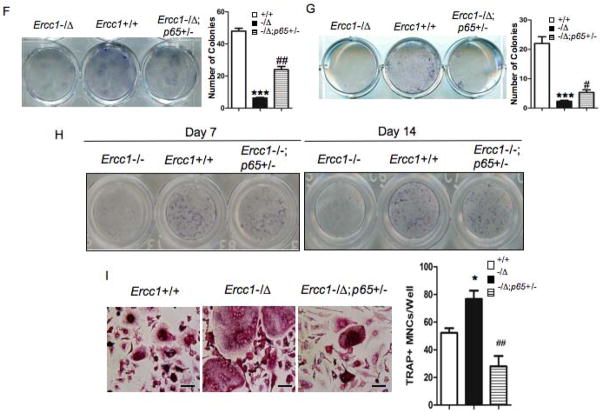
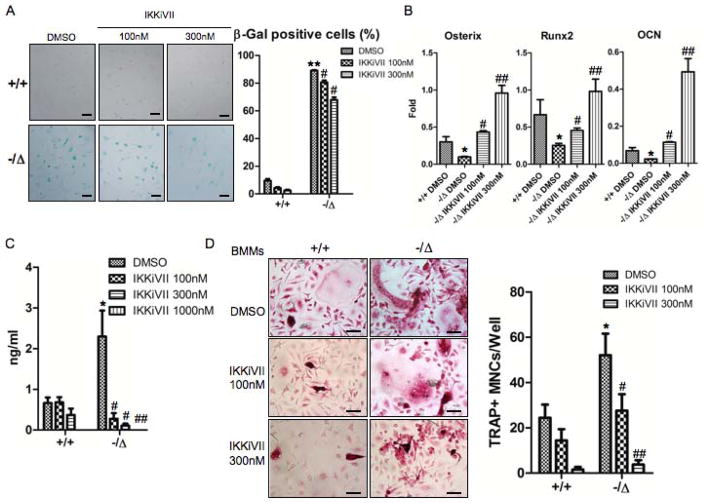
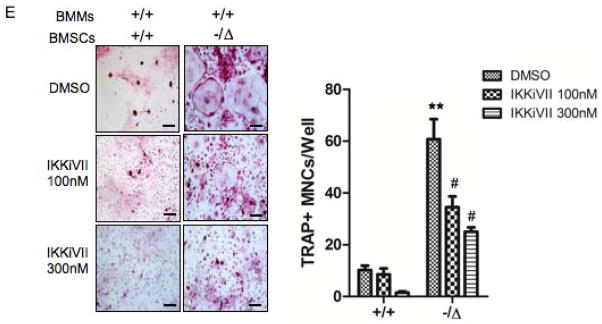
Advanced age is one of the most important risk factors for osteoporosis. Accumulation of oxidative DNA damage has been proposed to contribute to age-related deregulation of osteoblastic and osteoclastic cells. Excision repair cross complementary group 1-xeroderma pigmentosum group F (ERCC1-XPF) is an evolutionarily conserved structure-specific endonuclease that is required for multiple DNA repair pathways. Inherited mutations affecting expression of ERCC1-XPF cause a severe progeroid syndrome in humans, including early onset of osteopenia and osteoporosis, or anomalies in skeletal development. Herein, we used progeroid ERCC1-XPF-deficient mice, including Ercc1-null (Ercc1(-/-)) and hypomorphic (Ercc1(-/Δ)) mice, to investigate the mechanism by which DNA damage leads to accelerated bone aging. Compared to their wild-type littermates, both Ercc1(-/-) and Ercc1(-/Δ) mice display severe, progressive osteoporosis caused by reduced bone formation and enhanced osteoclastogenesis. ERCC1 deficiency leads to atrophy of osteoblastic progenitors in the bone marrow stromal cell (BMSC) population. There is increased cellular senescence of BMSCs and osteoblastic cells, as characterized by reduced proliferation, accumulation of DNA damage, and a senescence-associated secretory phenotype (SASP). This leads to enhanced secretion of inflammatory cytokines known to drive osteoclastogenesis, such as interleukin-6 (IL-6), tumor necrosis factor α (TNFα), and receptor activator of NF-κB ligand (RANKL), and thereby induces an inflammatory bone microenvironment favoring osteoclastogenesis. Furthermore, we found that the transcription factor NF-κB is activated in osteoblastic and osteoclastic cells of the Ercc1 mutant mice. Importantly, we demonstrated that haploinsufficiency of the p65 NF-κB subunit partially rescued the osteoporosis phenotype of Ercc1(-/Δ) mice. Finally, pharmacological inhibition of the NF-κB signaling via an I-κB kinase (IKK) inhibitor reversed cellular senescence and SASP in Ercc1(-/Δ) BMSCs. These results demonstrate that DNA damage drives osteoporosis through an NF-κB-dependent mechanism. Therefore, the NF-κB pathway represents a novel therapeutic target to treat aging-related bone disease.
Copyright © 2013 American Society for Bone and Mineral Research.
Conflict of interest statement
All authors state that they have no conflicts of interest.
Figures
References
-
- Cheung CL, Xiao SM, Kung AW. Genetic epidemiology of age-related osteoporosis and its clinical applications. Nat Rev Rheumatol. 2010;6(9):507–17. - PubMed
-
- Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531–57. - PubMed
-
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R21 CA161150/CA/NCI NIH HHS/United States
- P30 AG024827/AG/NIA NIH HHS/United States
- R01DE017439/DE/NIDCR NIH HHS/United States
- ES016114/ES/NIEHS NIH HHS/United States
- R21 AG033907/AG/NIA NIH HHS/United States
- R21CA161150/CA/NCI NIH HHS/United States
- AG024827/AG/NIA NIH HHS/United States
- R01 ES016114/ES/NIEHS NIH HHS/United States
- R01 AR055208/AR/NIAMS NIH HHS/United States
- U01 NS058451/NS/NINDS NIH HHS/United States
- P30 CA047904/CA/NCI NIH HHS/United States
- P30CA047904/CA/NCI NIH HHS/United States
- P30AG024827/AG/NIA NIH HHS/United States
- R01AR055208/AR/NIAMS NIH HHS/United States
- AR051456/AR/NIAMS NIH HHS/United States
- R01 AR051456/AR/NIAMS NIH HHS/United States
- R01 DE017439/DE/NIDCR NIH HHS/United States
- AG033907/AG/NIA NIH HHS/United States
- NS058451/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
