

Calcium cycling proteins and heart failure: mechanisms and therapeutics
- PMID: 23281409
- PMCID: PMC3533269
- DOI: 10.1172/JCI62834
Calcium cycling proteins and heart failure: mechanisms and therapeutics
Abstract
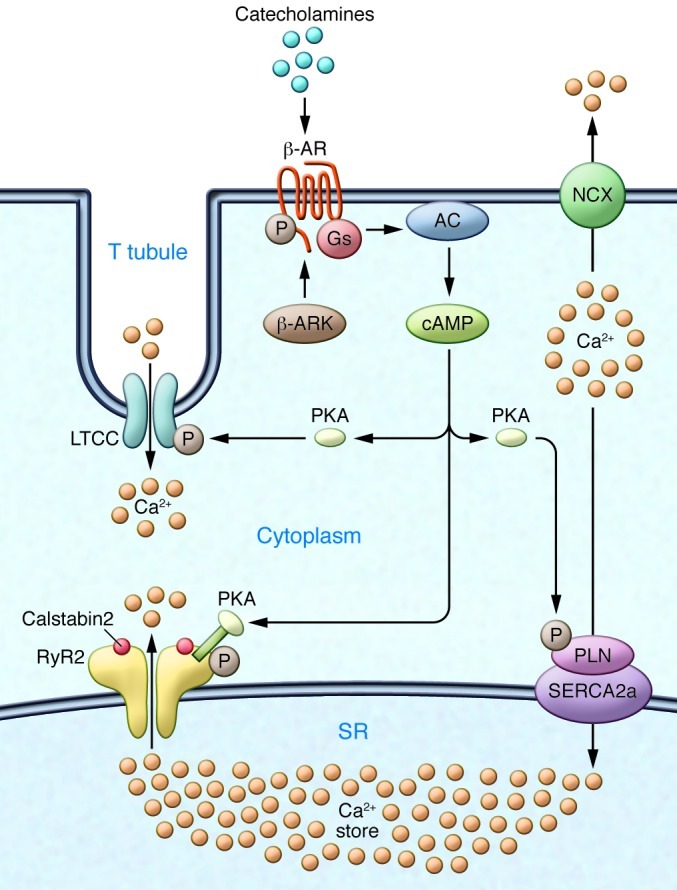
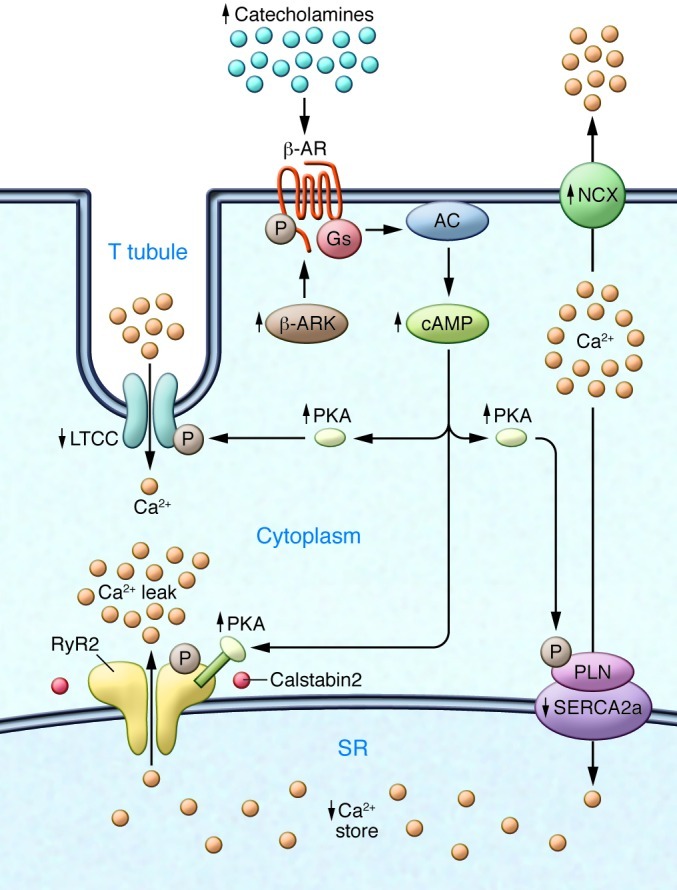
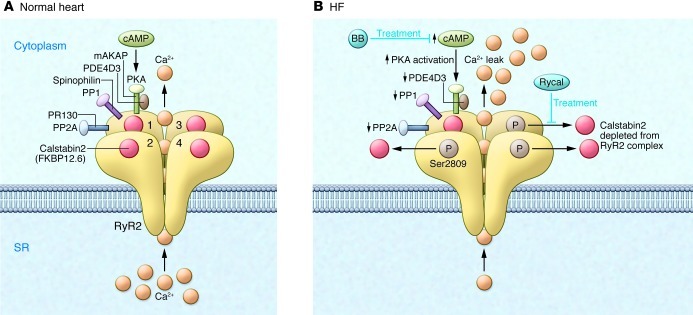
Ca2+-dependent signaling is highly regulated in cardiomyocytes and determines the force of cardiac muscle contraction. Ca2+ cycling refers to the release and reuptake of intracellular Ca2+ that drives muscle contraction and relaxation. In failing hearts, Ca2+ cycling is profoundly altered, resulting in impaired contractility and fatal cardiac arrhythmias. The key defects in Ca2+ cycling occur at the level of the sarcoplasmic reticulum (SR), a Ca2+ storage organelle in muscle. Defects in the regulation of Ca2+ cycling proteins including the ryanodine receptor 2, cardiac (RyR2)/Ca2+ release channel macromolecular complexes and the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a)/phospholamban complex contribute to heart failure. RyR2s are oxidized, nitrosylated, and PKA hyperphosphorylated, resulting in "leaky" channels in failing hearts. These leaky RyR2s contribute to depletion of Ca2+ from the SR, and the leaking Ca2+ depolarizes cardiomyocytes and triggers fatal arrhythmias. SERCA2a is downregulated and phospholamban is hypophosphorylated in failing hearts, resulting in impaired SR Ca2+ reuptake that conspires with leaky RyR2 to deplete SR Ca2+. Two new therapeutic strategies for heart failure (HF) are now being tested in clinical trials: (a) fixing the leak in RyR2 channels with a novel class of Ca2+-release channel stabilizers called Rycals and (b) increasing expression of SERCA2a to improve SR Ca2+ reuptake with viral-mediated gene therapy. There are many potential opportunities for additional mechanism-based therapeutics involving the machinery that regulates Ca2+ cycling in the heart.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
