

Examination of the regulatory frameworks applicable to biologic drugs (including stem cells and their progeny) in Europe, the U.S., and Australia: part II--a method of software documentary analysis
- PMID: 23283552
- PMCID: PMC3659673
- DOI: 10.5966/sctm.2012-0038
Examination of the regulatory frameworks applicable to biologic drugs (including stem cells and their progeny) in Europe, the U.S., and Australia: part II--a method of software documentary analysis
Abstract
A wide range of regulatory standards applicable to production and use of tissues, cells, and other biologics (or biologicals), as advanced therapies, indicates considerable interest in the regulation of these products. The objective of this study was to analyze and compare high-tier documents within the Australian, European, and U.S. biologic drug regulatory environments using qualitative methodology. Eighteen high-tier documents from the European Medicines Agency (EMA), U.S. Food and Drug Administration (FDA), and Therapeutic Goods Administration (TGA) regulatory frameworks were subject to automated text analysis. Selected documents were consistent with the legal requirements for manufacturing and use of biologic drugs in humans and fall into six different categories. Concepts, themes, and their co-occurrence were identified and compared. The most frequent concepts in TGA, FDA, and EMA frameworks were "biological," "product," and "medicinal," respectively. This was consistent with the previous manual terminology search. Good Manufacturing Practice documents, across frameworks, identified "quality" and "appropriate" as main concepts, whereas in Good Clinical Practice (GCP) documents it was "clinical," followed by "trial," "subjects," "sponsor," and "data." GCP documents displayed considerably higher concordance between different regulatory frameworks, as demonstrated by a smaller number of concepts, similar size, and similar distance between them. Although high-tier documents often use different terminology, they share concepts and themes. This paper may be a modest contribution to the recognition of similarities and differences between analyzed regulatory documents. It may also fill the literature gap and provide some foundation for future comparative research of biologic drug regulations on a global level.
Figures
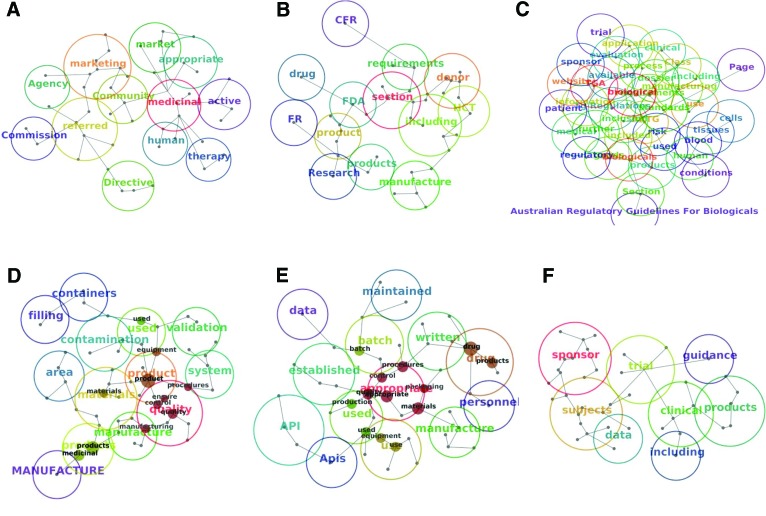

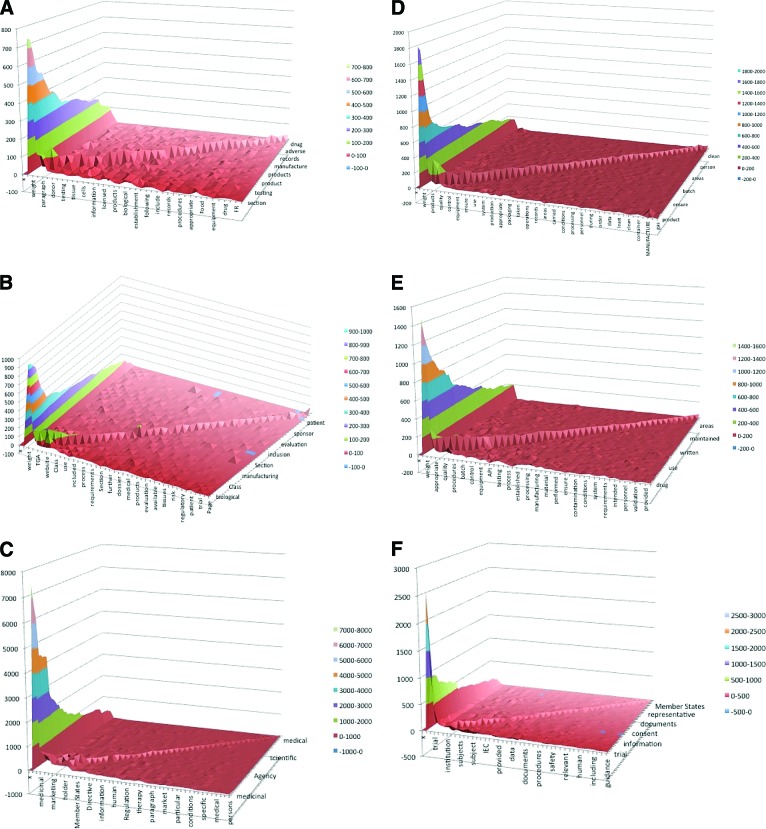
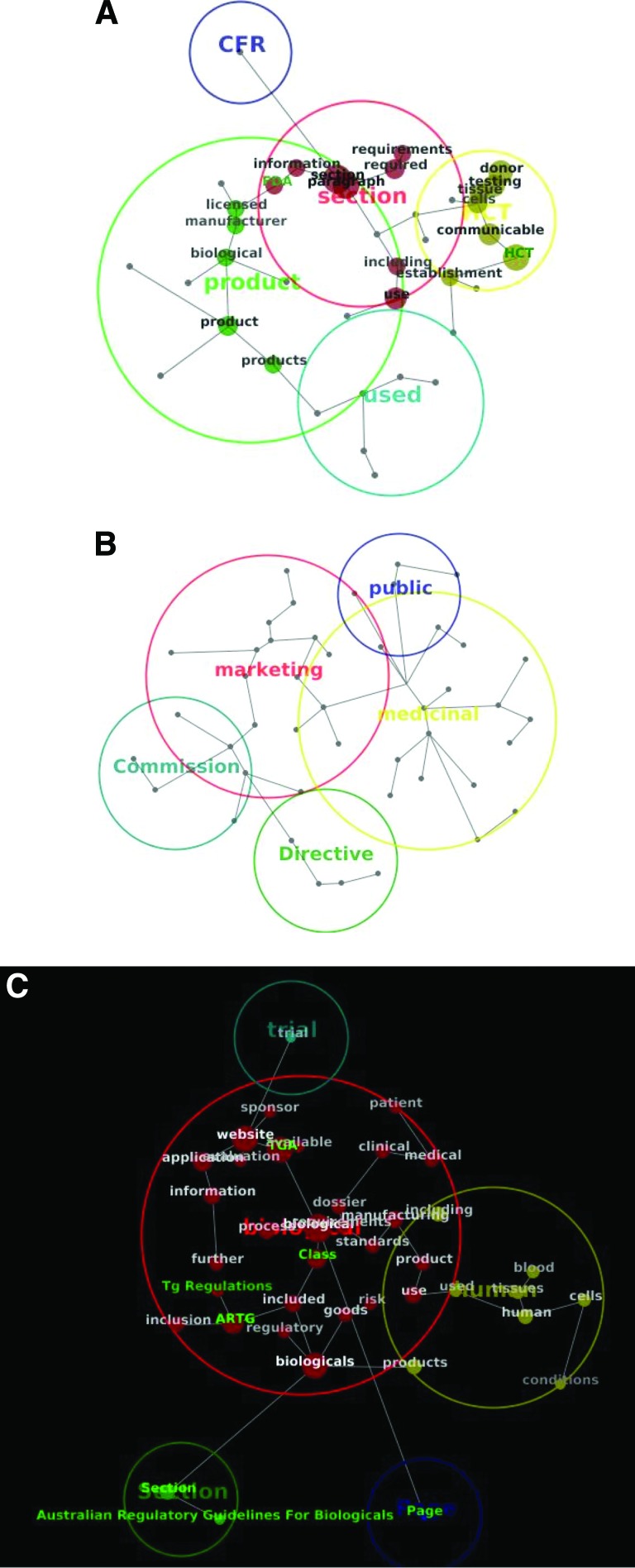
References
-
- Halme DG, Kessler DA. FDA regulation of stem-cell-based therapies. N Engl J Med. 2006;355:1730–1735. - PubMed
-
- FDA Guidance for industry: PAT—A framework for innovative pharmaceutical development, manufacturing, and quality assurance. 2004. [Accessed January 13, 2011]. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformati....
-
- The Financing of Biopharmaceutical Product Development in Europe. Final Report. European Commission, Enterprise and Industry. 2009. Oct, [Accessed December 5, 2011]. Available at http://ec.europa.eu/enterprise/sectors/biotechnology/files/docs/financin....
-
- Advancing Regulatory Science at FDA, Strategic Plan: August 2011. Food and Drug Administration, U.S. Department of Health and Human Services. [Accessed December 5, 2011]. Available at http://www.fda.gov/regulatoryscience.
-
- Implementing the European Medicines Agency's Road Map to 2015: The Agency's Contribution to Science, Medicine, Health—From Vision to Reality. EMA/MB/550544/2011. 2011. Oct 6, [Accessed November 22, 2011]. Available at http://www.ema.europa.eu.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
