

Epoxyeicosatrienoic acids (EETs) regulate epithelial sodium channel activity by extracellular signal-regulated kinase 1/2 (ERK1/2)-mediated phosphorylation
- PMID: 23283969
- PMCID: PMC3576126
- DOI: 10.1074/jbc.M112.407981
Epoxyeicosatrienoic acids (EETs) regulate epithelial sodium channel activity by extracellular signal-regulated kinase 1/2 (ERK1/2)-mediated phosphorylation
Abstract
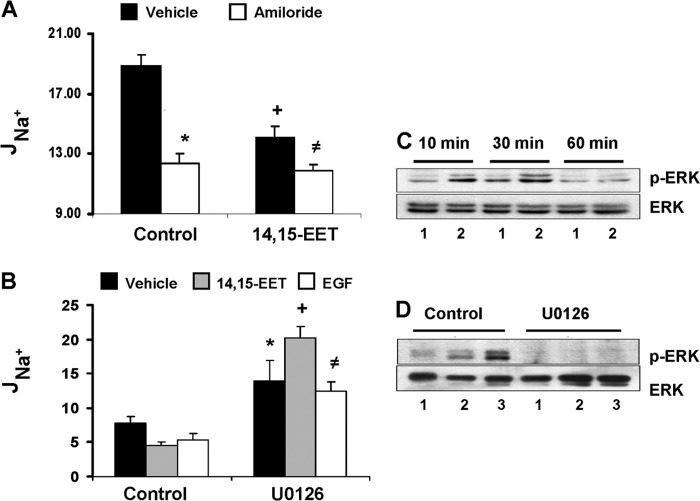
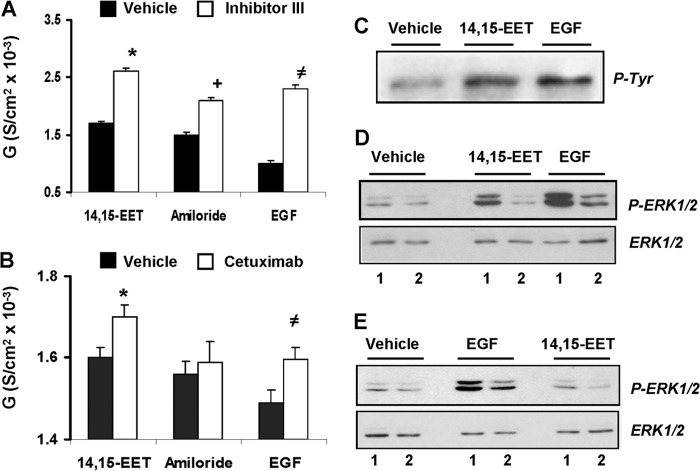
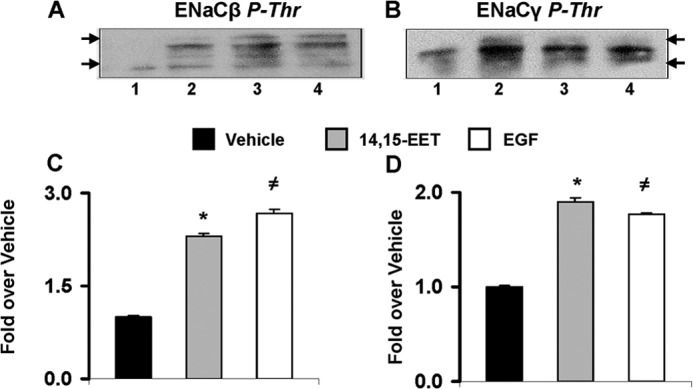
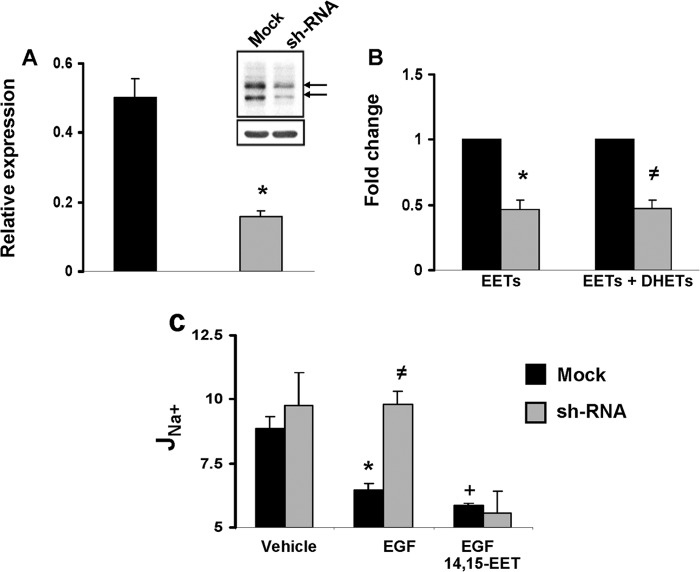
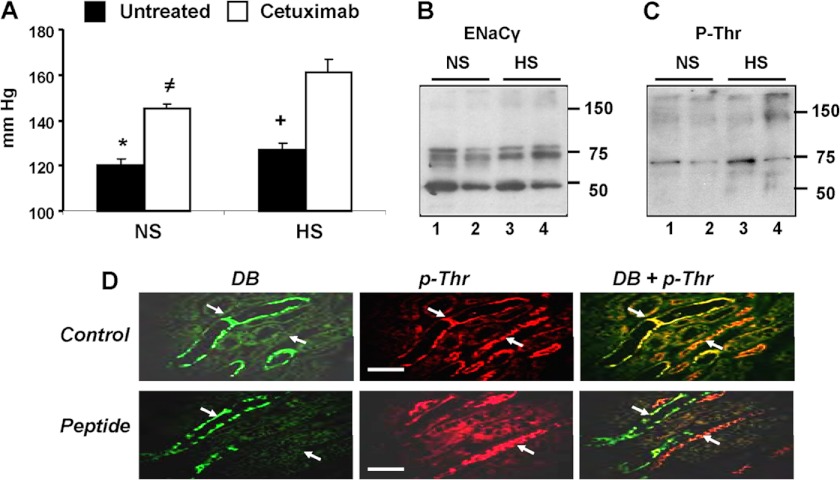
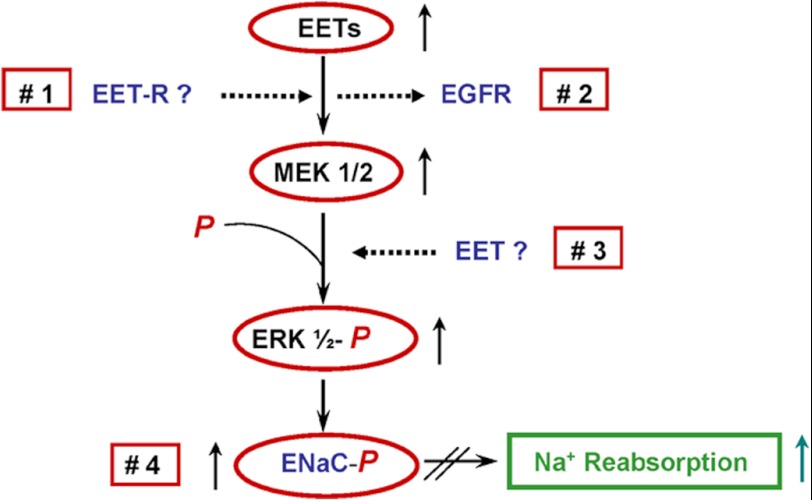
The epithelial sodium channel (ENaC) participates in the regulation of plasma sodium and volume, and gain of function mutations in the human channel cause salt-sensitive hypertension. Roles for the arachidonic acid epoxygenase metabolites, the epoxyeicosatrienoic acids (EETs), in ENaC activity have been identified; however, their mechanisms of action remain unknown. In polarized M1 cells, 14,15-EET inhibited amiloride-sensitive apical to basolateral sodium transport as effectively as epidermal growth factor (EGF). The EET effects were associated with increased threonine phosphorylation of the ENaC β and γ subunits and abolished by inhibitors of (a) mitogen-activated protein kinase/extracellular signal-regulated kinase kinase/extracellular signal regulated kinases 1 and 2 (MEK/ERK1/2) and (b) EGF receptor signaling. CYP2C44 epoxygenase knockdown blunted the sodium transport effects of EGF, and its 14,15-EET metabolite rescued the knockdown phenotype. The relevance of these findings is indicated by (a) the hypertension that results in mice administered cetuximab, an inhibitor of EGF receptor binding, and (b) immunological data showing an association between the pressure effects of cetuximab and reductions in ENaCγ phosphorylation. These studies (a) identify an ERK1/2-dependent mechanism for ENaC inhibition by 14,15-EET, (b) point to ENaC as a proximal target for EET-activated ERK1/2 mitogenic kinases, (c) characterize a mechanistic commonality between EGF and epoxygenase metabolites as ENaC inhibitors, and (d) suggest a CYP2C epoxygenase-mediated pathway for the regulation of distal sodium transport.
Figures
Similar articles
-
Arachidonic acid monooxygenase: Genetic and biochemical approaches to physiological/pathophysiological relevance.Prostaglandins Other Lipid Mediat. 2015 Jul;120:40-9. doi: 10.1016/j.prostaglandins.2015.05.004. Epub 2015 May 15. Prostaglandins Other Lipid Mediat. 2015. PMID: 25986599 Free PMC article. Review.
-
The Cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt.J Biol Chem. 2014 Feb 14;289(7):4377-86. doi: 10.1074/jbc.M113.508416. Epub 2013 Dec 24. J Biol Chem. 2014. PMID: 24368771 Free PMC article.
-
Cyp2c44 epoxygenase in the collecting duct is essential for the high K+ intake-induced antihypertensive effect.Am J Physiol Renal Physiol. 2014 Aug 15;307(4):F453-60. doi: 10.1152/ajprenal.00123.2014. Epub 2014 Jun 25. Am J Physiol Renal Physiol. 2014. PMID: 24966089 Free PMC article.
-
Transfection of an active cytochrome P450 arachidonic acid epoxygenase indicates that 14,15-epoxyeicosatrienoic acid functions as an intracellular second messenger in response to epidermal growth factor.J Biol Chem. 1999 Feb 19;274(8):4764-9. doi: 10.1074/jbc.274.8.4764. J Biol Chem. 1999. PMID: 9988714
-
Role of cytochrome P450 epoxygenase in regulating renal membrane transport and hypertension.Curr Opin Nephrol Hypertens. 2013 Mar;22(2):163-9. doi: 10.1097/MNH.0b013e32835d911e. Curr Opin Nephrol Hypertens. 2013. PMID: 23302865 Free PMC article. Review.
Cited by
-
Midkine Regulates BP through Cytochrome P450-Derived Eicosanoids.J Am Soc Nephrol. 2015 Aug;26(8):1806-15. doi: 10.1681/ASN.2013121259. Epub 2014 Nov 5. J Am Soc Nephrol. 2015. PMID: 25377079 Free PMC article.
-
Eicosanoid-Regulated Myeloid ENaC and Isolevuglandin Formation in Human Salt-Sensitive Hypertension.Hypertension. 2024 Mar;81(3):516-529. doi: 10.1161/HYPERTENSIONAHA.123.21285. Epub 2023 Sep 7. Hypertension. 2024. PMID: 37675576 Free PMC article.
-
Epidermal growth factors in the kidney and relationship to hypertension.Am J Physiol Renal Physiol. 2013 Jul 1;305(1):F12-20. doi: 10.1152/ajprenal.00112.2013. Epub 2013 May 1. Am J Physiol Renal Physiol. 2013. PMID: 23637204 Free PMC article. Review.
-
Arachidonic acid monooxygenase: Genetic and biochemical approaches to physiological/pathophysiological relevance.Prostaglandins Other Lipid Mediat. 2015 Jul;120:40-9. doi: 10.1016/j.prostaglandins.2015.05.004. Epub 2015 May 15. Prostaglandins Other Lipid Mediat. 2015. PMID: 25986599 Free PMC article. Review.
-
Modulation of K(Ca)3.1 channels by eicosanoids, omega-3 fatty acids, and molecular determinants.PLoS One. 2014 Nov 5;9(11):e112081. doi: 10.1371/journal.pone.0112081. eCollection 2014. PLoS One. 2014. PMID: 25372486 Free PMC article.
References
-
- Schild L. (1996) The ENaC channel as the primary determinant of two human diseases: Liddle syndrome and pseudohypoaldosteronism. Nephrologie 17, 395–400 - PubMed
-
- Warnock D. G. (2001) Liddle syndrome: genetics and mechanism of Na+ channel defects. Am. J. Med. Sci. 322, 302–307 - PubMed
-
- Rossier B. C., Schild L. (2008) Epithelial sodium channel. Mendelian versus essential hypertension. Hypertension 52, 595–600 - PubMed
-
- Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J. D., Rossier B. C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
