

A new isolation with migration model along complete genomes infers very different divergence processes among closely related great ape species
- PMID: 23284294
- PMCID: PMC3527290
- DOI: 10.1371/journal.pgen.1003125
A new isolation with migration model along complete genomes infers very different divergence processes among closely related great ape species
Abstract
We present a hidden Markov model (HMM) for inferring gradual isolation between two populations during speciation, modelled as a time interval with restricted gene flow. The HMM describes the history of adjacent nucleotides in two genomic sequences, such that the nucleotides can be separated by recombination, can migrate between populations, or can coalesce at variable time points, all dependent on the parameters of the model, which are the effective population sizes, splitting times, recombination rate, and migration rate. We show by extensive simulations that the HMM can accurately infer all parameters except the recombination rate, which is biased downwards. Inference is robust to variation in the mutation rate and the recombination rate over the sequence and also robust to unknown phase of genomes unless they are very closely related. We provide a test for whether divergence is gradual or instantaneous, and we apply the model to three key divergence processes in great apes: (a) the bonobo and common chimpanzee, (b) the eastern and western gorilla, and (c) the Sumatran and Bornean orang-utan. We find that the bonobo and chimpanzee appear to have undergone a clear split, whereas the divergence processes of the gorilla and orang-utan species occurred over several hundred thousands years with gene flow stopping quite recently. We also apply the model to the Homo/Pan speciation event and find that the most likely scenario involves an extended period of gene flow during speciation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
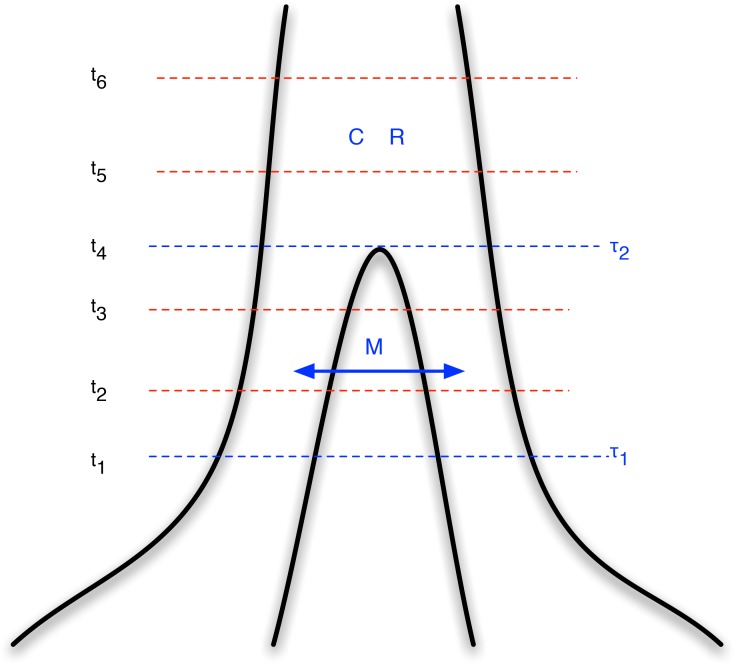
 , and that these two populations exchanged genes with migration rate
, and that these two populations exchanged genes with migration rate  until a later time,
until a later time,  , where gene flow stopped. The coalescence process in this model is parameterized with a coalescence rate (inverse of the effective population size),
, where gene flow stopped. The coalescence process in this model is parameterized with a coalescence rate (inverse of the effective population size),  , and a recombination rate,
, and a recombination rate,  . The model is translated into a finite-state hidden Markov model by discretizing time into time intervals with break points
. The model is translated into a finite-state hidden Markov model by discretizing time into time intervals with break points  .
.
 , the initial population split,
, the initial population split,  , and the migration rate,
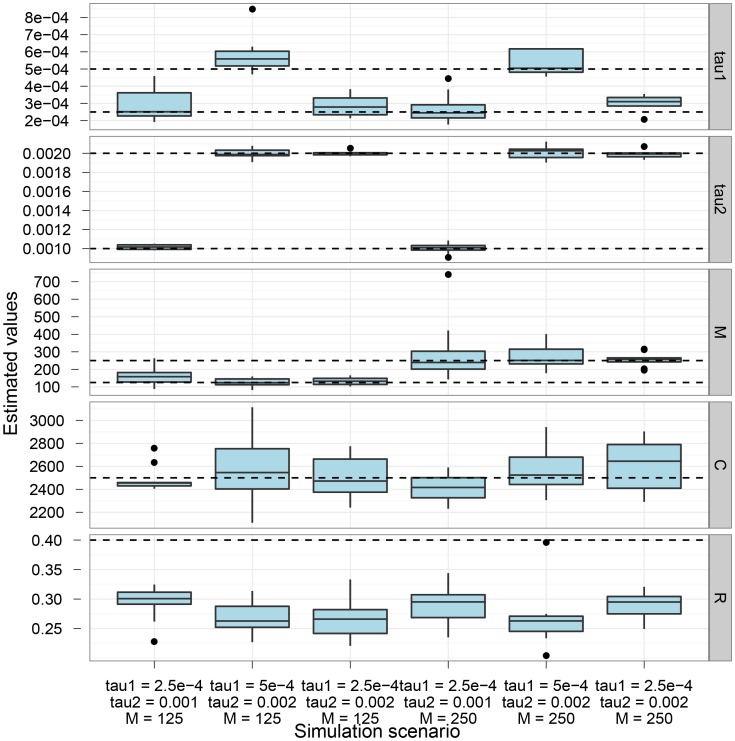
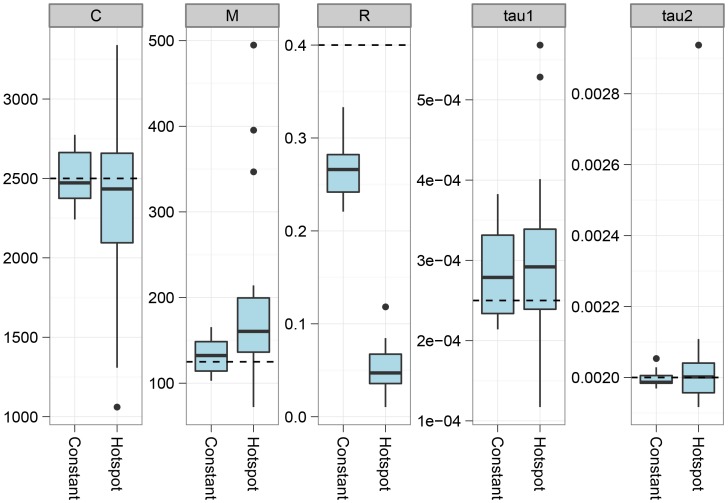
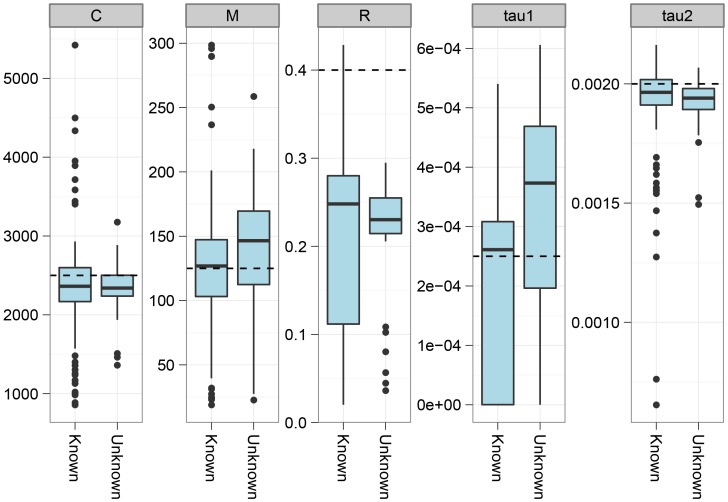
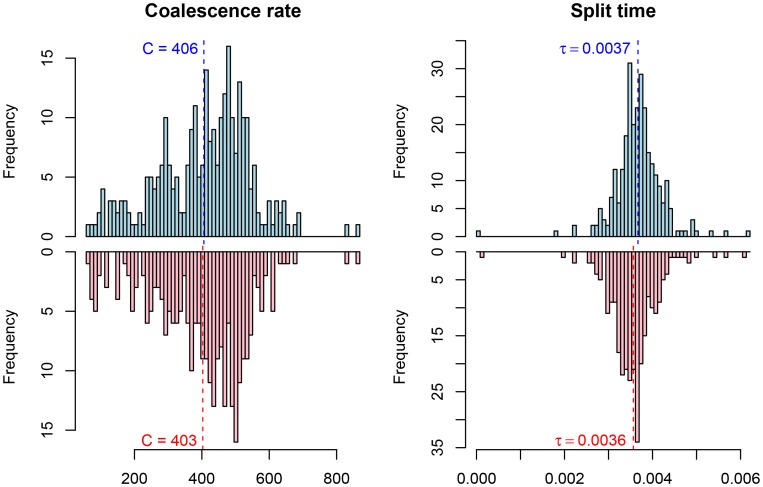
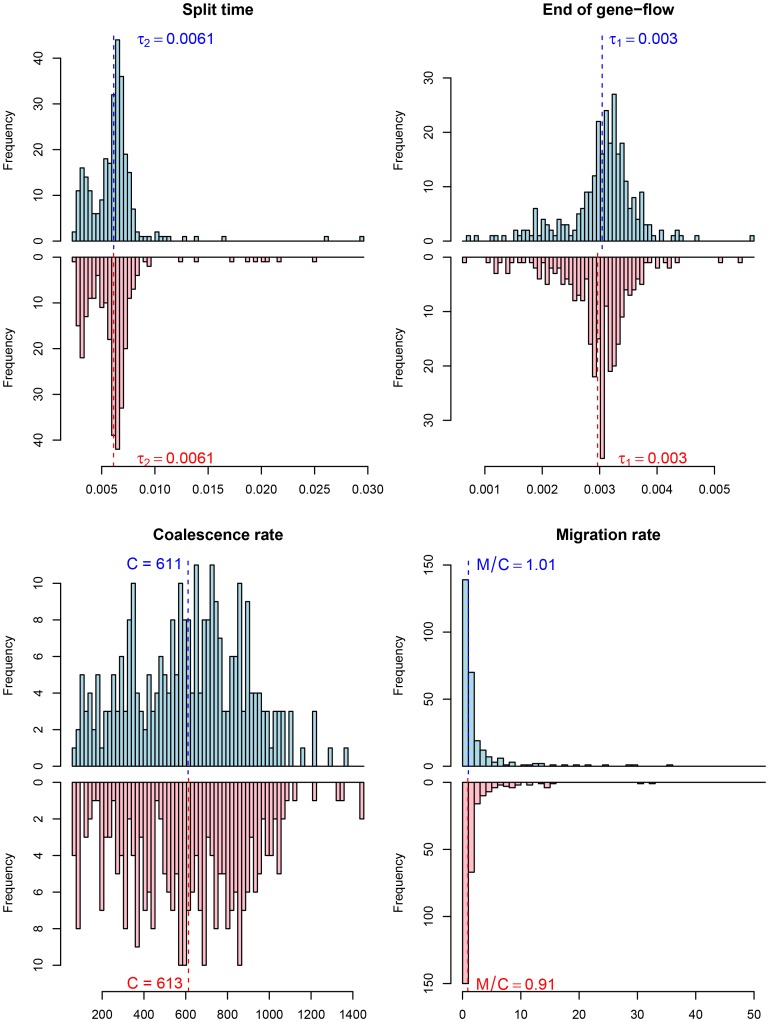
, and the migration rate,  , varies between scenarios. For each simulation scenario, 10 independent data sets were generated and analyzed. The dashed horizontal lines indicate the simulated values for the five parameters. The recombination rate is consistently under-estimated while the remaining parameters are well recovered.
, varies between scenarios. For each simulation scenario, 10 independent data sets were generated and analyzed. The dashed horizontal lines indicate the simulated values for the five parameters. The recombination rate is consistently under-estimated while the remaining parameters are well recovered.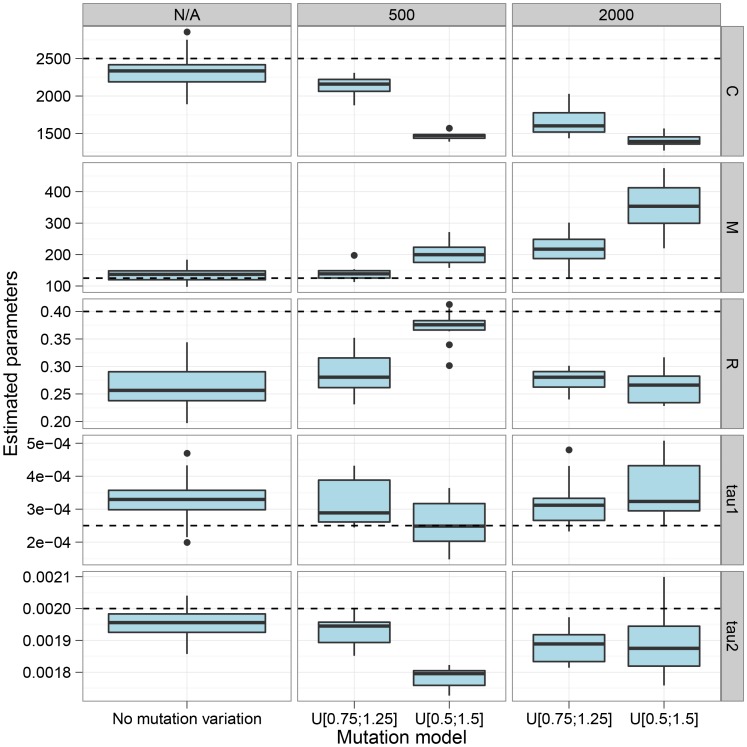
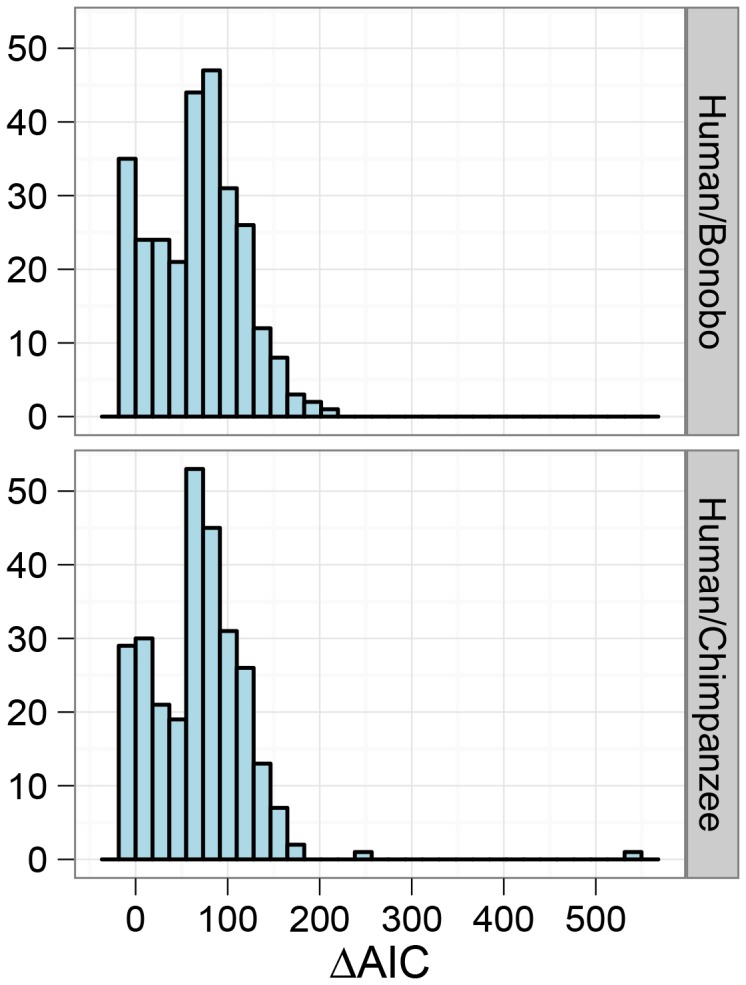
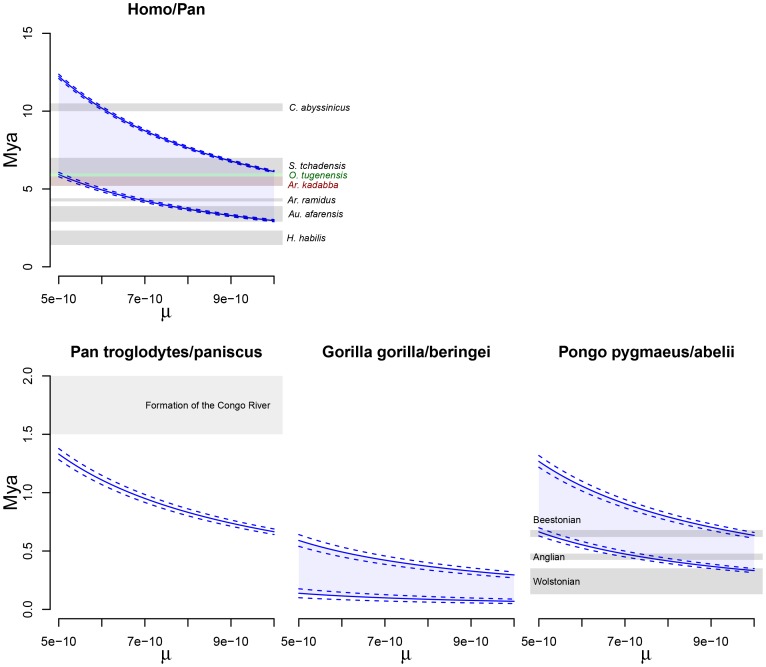
 , ranging from
, ranging from  to
to  . The solid lines show mean estimates while the dashed lines the 95% confidence interval (
. The solid lines show mean estimates while the dashed lines the 95% confidence interval ( SEM). The Homo/Pan slit is annotated with key fossils, the chimpanzee/bonobo split with the formation of the Congo River, and the orang-utan split with glacial period where sea level was low and migration between orang-utans possible.
SEM). The Homo/Pan slit is annotated with key fossils, the chimpanzee/bonobo split with the formation of the Congo River, and the orang-utan split with glacial period where sea level was low and migration between orang-utans possible.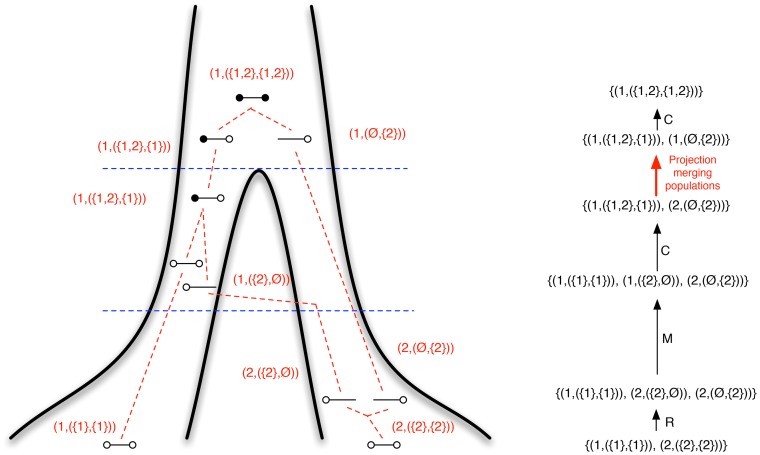
References
-
- Burgess R, Yang Z (2008) Estimation of hominoid ancestral population sizes under bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol Biol Evol 25: 1979–1994. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
