

FastUniq: a fast de novo duplicates removal tool for paired short reads
- PMID: 23284954
- PMCID: PMC3527383
- DOI: 10.1371/journal.pone.0052249
FastUniq: a fast de novo duplicates removal tool for paired short reads
Abstract
The presence of duplicates introduced by PCR amplification is a major issue in paired short reads from next-generation sequencing platforms. These duplicates might have a serious impact on research applications, such as scaffolding in whole-genome sequencing and discovering large-scale genome variations, and are usually removed. We present FastUniq as a fast de novo tool for removal of duplicates in paired short reads. FastUniq identifies duplicates by comparing sequences between read pairs and does not require complete genome sequences as prerequisites. FastUniq is capable of simultaneously handling reads with different lengths and results in highly efficient running time, which increases linearly at an average speed of 87 million reads per 10 minutes. FastUniq is freely available at http://sourceforge.net/projects/fastuniq/.
Conflict of interest statement
Figures
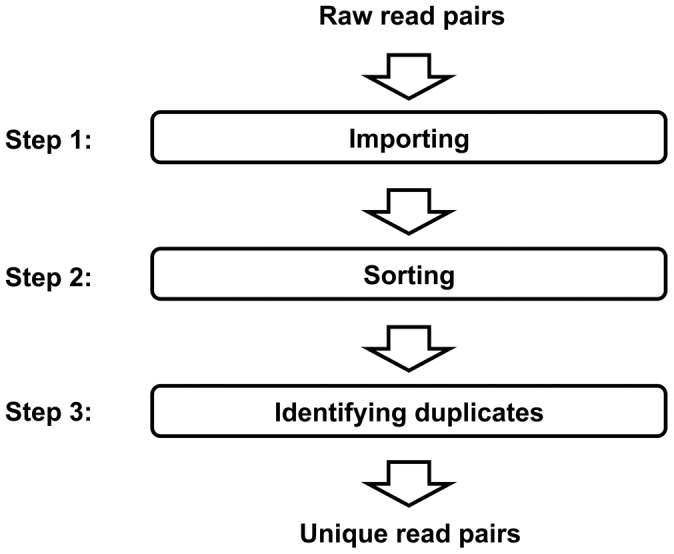
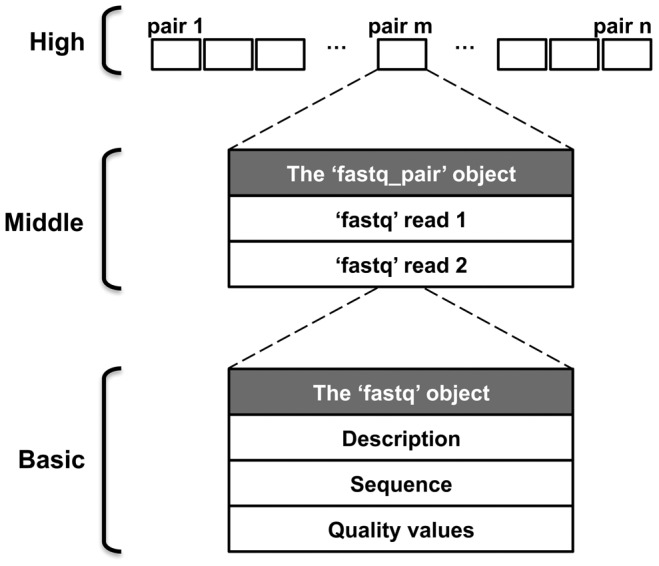
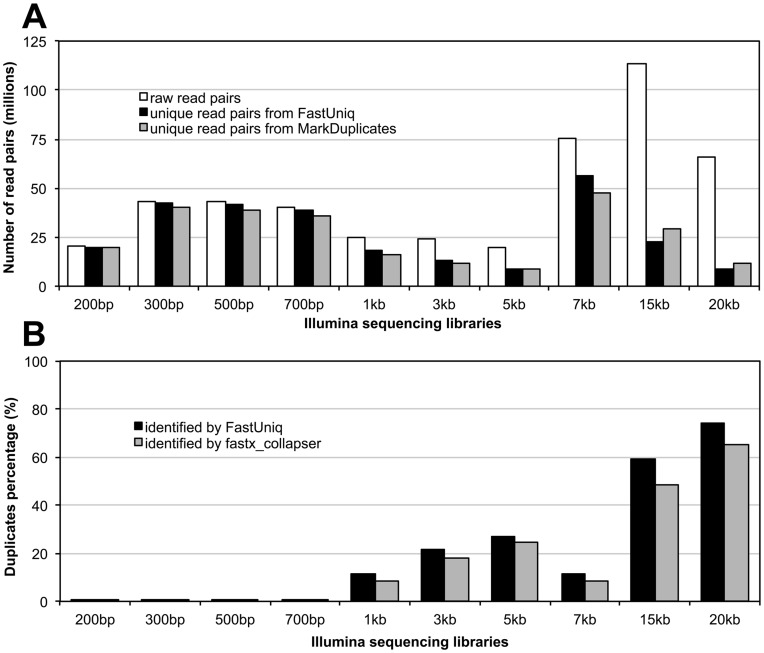
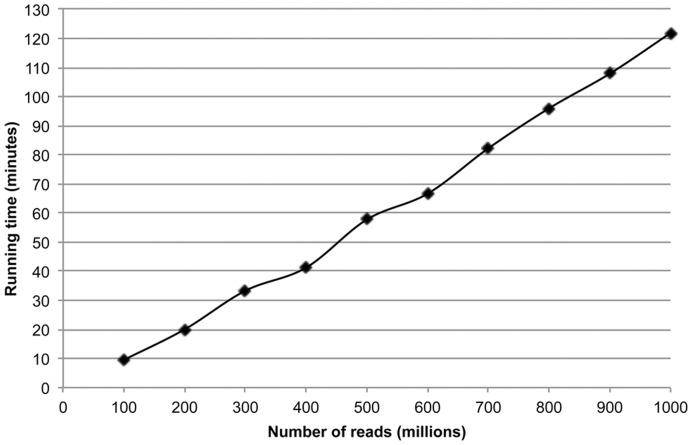
References
-
- Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, et al. (2011) Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476: 320–323. - PubMed
-
- Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, et al. (2007) High-throughput oncogene mutation profiling in human cancer. Nat Genet 39: 347–351. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
